Les anomalies chromosomiques désignent l’aneuploïdie avec la perte ou l’ajout de chromosomes entiers, mais aussi d’autres réarrangements chromosomiques comme la délétion, la duplication ou la translocation de morceaux de chromosomes (Béri-Dexheimer, 2005). Ces anomalies entraînent des conséquences variées tels que les retards de développement, l’autisme et d’autres anomalies congénitales (Shinawi et Cheung, 2008). L’hybridation génomique comparative (HGC) est une méthode utilisée pour la détection de ces pathologies, notamment au stade fœtal (Jonveaux, 2010), et se révèle être prometteuse dans la recherche sur le cancer.
II)Principe
L’hybridation génomique comparative se base sur les déséquilibres génomiques pour diagnostiquer ces anomalies chromosomiques. La technique est actuellement utilisée sur puce à ADN pour plus d’efficacité et de précision, diminuant ainsi la taille des déséquilibres détectables. La technique repose sur l’hybridation d’ADN de référence et d’ADN à tester (Shinawi, et Cheung, 2008).
III)Mode opératoire (Figure 1)
La puce d’hybridation génomique comparative est recouverte de séquences d’ADN cibles de longueur variable en fonction de la précision voulue. Deux sortes de sondes sont préparées : l’ADN à tester marqué par la Cyanine 3 et l’ADN de référence marqué par la Cyanine 5. De l’ADN Cot-1 est ajouté pour éviter l'hybridation de séquences répétées. Les deux sondes sont ensuite déposées sur la lame pour l’hybridation. La puce est ensuite lavée et lue à l’aide d’un scanner laser. Les intensités de fluorescence des ADN marqués à la Cyanine 3 et à la Cyanine 5 sont comparées à l’aide d’une représentation graphique (Béri-Dexheimer et al, 2007).
Figure 1 : Principe de l'hybridation génomique comparative sur microréseau d'ADN (Béri-Dexheimeret al, 2007).
IV)Présentation des résultats
Après hybridation, la fluorescence est capturée à l’aide d’une caméra (HGC métaphasique) ou d’un scanner laser pour l’hybridation génomique comparative sur microréseau cette image montre déjà des premiers résultats. En effet, les points rouges indiquent une perte de l’ADN test, les points verts un gain de l’ADN test et les points jaunes indiquent la présence d’une quantité équivalente entre l’ADN test et l’ADN témoin. Pour une analyse quantitative, un logiciel calcule les rapports de fluorescence entre les deux ADN, et représente les résultats sous forme de graphique (Jonveaux, 2010) (Figure 2). L’analyse est réalisée pour l’ensemble des cibles présentes sur la lame. Chaque point du graphique correspond à une cible dont la cartographie physique sur le chromosome est connue permettantainsi de quantifier la taille de l’anomalie et d’apprécier les gènesimpliqués dans le remaniement (Béri-Dexheimeret al, 2007).
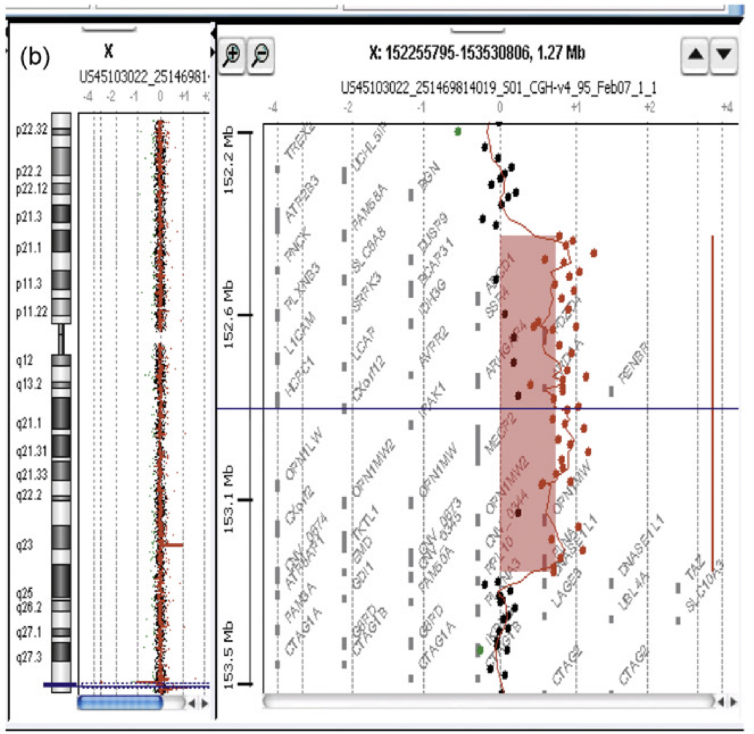
Figure 2 : Exemple d’un rapport d’une puce à oligonucléotides de la firme Agilent Technologies avec identification d’un gain en Xq28 (Jonveaux, 2010).
À gauche est représenté le pictogramme du chromosome X avec une déviation reconnue par le logiciel dans la partie terminale du bras long du chromosome X (ligne bleue). À droite est représenté un zoom de la région en déséquilibre avec chaque point rouge du graphique correspondant à une cible.
V)Interprétation des résultats
L’interprétation quantitative des résultats de l’hybridation génomique comparative se fait par l’interprétation de la courbe de résultats (figure 2). En pratique, pour chaque cible étudiée,
un ratio de fluorescence égal à 1 correspond à un nombre de copies normales, c’est-à-dire deux,
entre 1,3 et 1,4 à un gain d’une copie,
et entre 0,6 et 0,7 à la perte d’une copie.
Pour des problèmes de représentation graphique, la plupart des auteurs utilisent le log 2 de ce ratio. Sur la figure 1, la position génomique de chaque cible est donnée en Mb par rapport au télomère du chromosome étudié. De plus, l’échelle des ratios a été modifiée et une déviation à +1 correspond à un gain, tandis qu’une déviation à -1 correspond à une perte. Ici, il s’agit d’un gain d’une taille de 751 Kb s’étendant sur la séquence de référence du génome humain des nucléotides 152 511 063 à 153 262 357.
VI)Intérêts et limites
La méthode d’hybridation génomique comparative a de nombreuses fois montré son efficacité, cependant, elle possède quelques limites. Premièrement, son incapacité à détecter les réarrangements de structure équilibrés, ce qui peut conduire à des erreurs dans l'identification des anomalies génétiques. De plus, cette méthode ne permet pas la détection des anomalies causées par des interruptions de gènes. Elle cause aussi une limitation dans la détection des anomalies de la ploïdie telles que la triploïdie, qui ne sont pas identifiables car les échantillons test et témoin contiennent la même proportion de lot haploïde par quantité d'ADN hybridée. Enfin, avec cette méthode, l’interprétation des variations numériques chromosomiques est complexe, et seule une collecte extensive de données cliniques et biologiques sur de grandes populations peut aider à clarifier le caractère pathogène de certaines de ces variations.
VII)Références bibliographiques
Béri-Dexheimer M. (2005). Hybridation génomique comparative en microréseau : évolutions techniques et place dans la stratégie diagnostique, expérience dans le cadre du retard mental. Sciences pharmaceutiques.
Béri-Dexheimer M., Bonnet C., Chambon P., Brochet K., Grégoire M.-J., Jonveaux P. (2007). L'hybridation génomique comparative sur microréseau d'ADN (puces à ADN) en pathologie chromosomique constitutionnelle, Pathologie Biologie, Volume 55, Issue 1, p.13-18.
Jonveaux P. (2010). Technique d’hybridation génomique comparative sur microréseau d’ADN et fœtopathologie, Archives de Pédiatrie, Volume 17, Issue 7, p.1119-1123.
Shinawi M., Cheung S. W. (2008). The array CGH and its clinical applications, Drug Discovery Today, Volume 13, Issues 17–18, p.760-770.