CRISPR-Cas9 est une technique d’édition du
génome développée par Emmanuelle Charpentier et Jennifer Doudna à partir de
2012 (4). Actuellement, ce système permet de supprimer une
mutation voire un gène entier, d’insérer un nouveau gène dans un génome ou
encore de créer des SNPs. Cette technique peut être utilisée pour faciliter un
large panel d’applications en génie génétique, en particulier pour la
manipulation ciblée du génome et les thérapies géniques.
II)Principe
La méthode repose sur la capacité de
certaines bactéries à résister aux virus grâce aux séquences CRISPR (Clustered
Regularly Interspersed Palindromic Repeats), qui sont transcrites en des ARN contenant
le complémentaire de l’ADN viral et qui se fixent à la nucléase Cas9.Lorsque
ce complexe Cas9-ARN reconnait la séquence complémentaire dans la cellule, il
le découpe. C’est cette capacité du complexe à identifier une séquence cible et
à la lyser qui fait de CRISPR-Cas9 un outil d’édition précis du génome (3).
L’activité nucléase de la protéine Cas9, lui
permet d’introduire des Double Stranded Breaks (DSBs), c’est-à-dire des
cassures franches sur les deux brins de la molécule d’ADN. Lorsque ces cassures
sont repérées par le système de réparation cellulaire, elles sont corrigées de
deux manières différentes.
D’abord, le Non Homologous End Joining
(NHEJ), mécanisme par lequel la cellule insère ou retire de manière aléatoire
des nucléotides au niveau de la cassure pour permettre de rabouter les deux
extrémités. Ce mécanisme est très imprécis, et tend à provoquer des mutations
de type Insertion-Deletion (Indels).
L’autre possibilité est la réparation par Homology
Directed Repair (HDR), mécanisme par lequel la cellule se base sur une
matrice d’ADN de même séquence, qui peut être l’autre allèle du gène concerné,
ou bien une matrice d’ADN fournie à la cellule, par exemple sous forme de single-stranded
oligonucleotides (ssODNs).
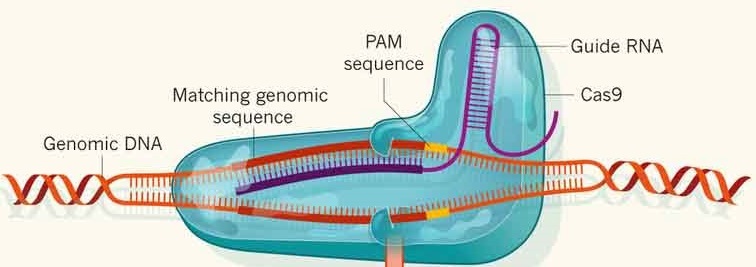
III)Mode opératoire
La méthode consiste à fournir à la cellule, soit directement la protéine
Cas9, soit sa séquence sous forme d’ADN plasmidique ou d’ARNm.
La protéine doit disposer d’un ARN guide (sgRNA), qui est une
chimère artificielle entre le crRNA (pour CRISPR) et le tracrRNA (pour trans-activating),
et qui lui sert notamment de modèle pour trouver la séquence d’intérêt sur
l’ADN de la cellule-cible. Cet ARN peut lui être fourni séparément par
transfection dans la cellule-cible, ou bien se trouver sur le même plasmide,
sous contrôle d’un promoteur. Cas9 balaie ensuite le génome de la cellule-cible
en ne s’arrêtant que lorsqu’elle rencontre un motif correspondant au PAM (Protospacer
Adjacent Motif), dont la séquence canonique, présente toutes les 8 bases
environ dans le génome humain, est 5’-NGG-3’. Si la séquence d’ADN en amont du
PAM correspond à la séquence-cible, Cas9 réalise une cassure double brin entre
le 3ième et le 4ième nucléotide en 5’ du 5’-NGG-3’.
De nombreuses versions modifiés de cas9 existent, avec une activité
nucléase sur un seul brin (nickase), sans activité nucléase (dCas9), avec un
PAM modifié (5’-YN-3’), se comportant comme un facteur de transcription, etc...
(1, 5)
Figure 1: Targeted genome
editing with RNA-guided Cas9 (2)
IV)Présentation des résultats
La présentation varie selon le type de
Cas9 utilisée et l’objectif de la manipulation. La mesure de l’activité d’une
protéine fluorescente peut permettre de mesurer l’efficacité d’une transfection
si l’on a transfecté une protéine-chimère, son ARNm ou bien un plasmide.
Pour démontrer l’efficacité de la
modification d’une séquence cible, on peut réaliser un séquençage type Sanger
de cette portion précise du génome. Pour s’assurer qu’aucune autre zone du
génome n’a été modifié par erreur (off-target), on pourra soit
rechercher les sites ayant une homologie forte avec la séquence cible et les
séquencer également, soit faire appel à une méthode de séquençage haut-débit
pour séquencer l’ensemble du génome.
V)Interprétation des résultats
Dans un premier temps, la mesure d’une
activité fluorescente, par exemple, dans la cellule-cible permet de valider la
transfection de Cas9. Cela permet d’éliminer les cellules non-transfectées du
champ d’étude. Enfin, le résultat d’un séquençage est analysé par alignement
avec soit la séquence de départ, soit la séquence souhaitée. La conclusion de
cet alignement est binaire : s’il est correct, la modification s’est
réalisée avec succès. Dans le cas contraire, la technique n’a pas été efficace.
VI)Intérêts et limites
Moins coûteuse et plus
rapide que les autres techniques d’édition du génome, CRISPR-Cas9 a été testée
sur des bactéries, des cellules végétales, animales et humaines. Le principal intérêt
de cette méthode est qu’elle facilement déclinable car il “suffit” de changer
la séquence guide du complexe pour l’adapter à une autre édition génomique.
Cependant, cette méthode
n’est pas fiable à 100% car une séquence complémentaire de la séquence guide
pourrait se trouver à un autre endroit du génome et il y aurait donc une
mutation sur une autre séquence. Cependant, cette limite n’en est plus vraiment
une car une alternative à Cas9 a été développée. En mutant un des deux domaines
nucléases de Cas9, les chercheurs ont créé une “nickase” qui ne réalise plus
qu’une seule coupure simple-brin. Avec une paire de nickases, chacun ayant une
séquence guide correspondant à un des brins, la spécificité de la mutation est
améliorée (6).
Enfin, des limites
éthiques à cette méthode en pleine expansion commencent à apparaitre et doivent
être discutées. En effet, la manipulation des cellules somatiques humaines
devient plus accessible, avec des risques d’eugénisme en conséquence. De plus
les mutations réalisées ne sont pas identifiables par un tiers contrairement
aux techniques actuelles. Le caractère génétiquement modifié d’un organisme
n’est donc plus vérifiable.
VII)Références bibliographiques
1. Bolukbasi, M. F.,
Gupta, A. & Wolfe, S. A. (2015) Creating and evaluating accurate CRISPR-Cas9 scalpels for genomic
surgery. Nature Methods13, 41–50
2. Charpentier, E. & Doudna, J. A. (2013) Biotechnology: Rewriting a genome. Nature495, 50–51
3. Hsu, P. D.,
Lander, E. S. & Zhang, F. (2014) Development and Applications of CRISPR-Cas9 for Genome Engineering.
Cell157, 1262–1278
4. Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna
J.A., Charpentier, E.(2012) A
Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science337, 816–821
5. Perez-Pinera, P., Kocak,
D. D., Vockley, C.M., Adler, A. F., Kabadi, A. M., Polstein, L. R., Thakore, P.
I., Glass, K. A., Ousterout, D. G., Leong, K. W., Guilak, F., Crawford, G. E., Reddy,
T. E, & Gersbach, C. A. (2013) RNA-guided gene activation by CRISPR-Cas9–based transcription
factors. Nature Methods10, 973–976
6. Ran, F.A., Hsu,
P.D., Lin, C.-Y., Gootenberg, J.S., Konermann, S., Trevino, A.E., Scott, D.A.,
Inoue, A., Matoba, S., Zhang, Y., Zhang, F. (2013) Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing
Specificity.Cell 154, 1380–1389