Le séquençage d'ARN (RNA-Seq) utilise les
capacités des méthodes de séquençage à haut débit pour fournir un aperçu du
transcriptome d'une cellule. Par rapport aux méthodes précédentes de Sanger (1) et de microarray (2), le RNA-Seq offre une
couverture beaucoup plus élevée et une plus grande résolution de la nature
dynamique du transcriptome. Au-delà de la quantification de l'expression des
gènes, les données générées par le RNA-Seq facilitent la découverte de nouveaux
transcrits, l'identification de transcrits issus d’épissage alternatif et la
détection de l'expression spécifique des allèles. Les améliorations récentes du
workflow du RNA-Seq, de la
préparation de l'échantillon à la construction de la bibliothèque jusqu'à
l'analyse des données, ont permis aux chercheurs d'élucider davantage la
complexité fonctionnelle de la transcription. En plus des transcrits d'ARN messager
(ARNm) polyadénylés, le RNA-Seq peut être appliqué pour étudier différentes
populations d'ARN, y compris l'ARN total, les pré-ARNm, et les ARN non codant,
tels que les microARN et les ARNc long. (3)
II)
Principe
La bibliothèque d'ADNc est générée par
transcription inverse comprenant des adaptateurs de séquençage spécifiques avec
des séquences « codes-barres ». Ensuite, les bibliothèques sont
regroupées sur des plaques, les flowcells,
et séquencées sur différentes machines (ex : Illumina). Le nombre envisagé
de lectures par bibliothèque dépend de l'organisme étudié et de la sensibilité
souhaitée.
Alors que la référence pour les génomes eucaryotes complexes (par exemple
humain, rat, souris) nécessite des lectures de 100-150 nt (sensibilité élevée) ou
20-30 nt lectures (faible sensibilité), une quantité de lecture 10 fois plus faible est
requise pour les bactéries. (4)
III)
Mode
opératoire
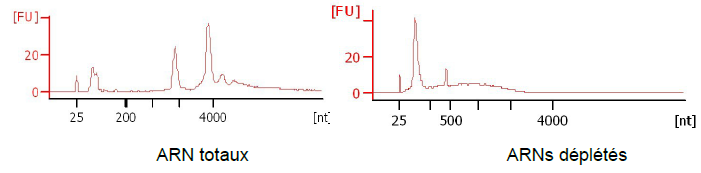
Une extraction des ARN totaux est réalisée.
Ces ARN sont ensuite contrôlés sur Bioanalyser (Agilent) pour vérifier leur
intégrité (figure 1).
Figure
1 : Comparaison des courbes d’ARN total et d’ARN déplétés (4)
Les ARNm matures sont ensuite extraits au
moyen de billes portant des séquences polyT auxquelles les queues polyA,
spécifiques des ARNm matures, se lient (figure 2).
Figure
2 : Purification des ARNm matures (5)
Les ARNm sont transcrits en ADNc double brin
par réverse transcription, et ceux-ci sont fragmentés chimiquement en fragments
d’environ 200 pb. Enfin, on lie des adaptateurs aux deux extrémités des
fragments d’ADNc double brin. Ces adaptateurs sont composés de séquences
orientées et spécifiques :
-
à l’échantillon (séquence
« Ech ») : ce sont des séquences codes-barres nécessaires au
multiplexage (car elles permettent de distinguer plusieurs échantillons sur une
même flowcell)
-
à la flowcell (séquences P1 et P2) : elles correspondent aux
cotés 3’ et 5’ et peuvent être utilisées pour une pré-amplification PCR. Elles
sont compatibles avec la flowcell où
sont fixés des oligonucléotides permettant le séquençage. (6)
Figure
3 : Organisation des ADNc finaux (7)
Figure 4 :
Workflow de la préparation d’une librairie d’ADNc pour le RNA-seq (8)
IV)
Présentation
des résultats
A l’issu
de la préparation de librairie une solution contenant les fragments d’ADNc à
séquencer est obtenue. Ces fragments sont ensuite déposés sur une plaque sur
laquelle sont fixés des oligonucléotides correspondants aux séquences P1 et P2
(voir partie mode opératoire). Cette plaque ou flowcell est ensuite placée dans un séquenceur (ex :
séquenceur Illumina).
V)
Interprétation
des résultats
La préparation de librairie en elle-même ne
fournit aucun résultat. Le séquençage des librairies permet la découverte de
nouveaux gènes ou de déterminer le niveau d’expression d’un gène donné.
VI)
Intérêts
et limites
Un intérêt majeur du RNA-seq est la
possibilité d’un multiplexage, à savoir une analyse simultanée de plusieurs
échantillons grâce aux séquences barcode.
Certaines manipulations au cours de la
construction de la bibliothèque compliquent l'analyse des résultats de RNA-seq.
Par exemple, de nombreuses « courtes lectures » peuvent être
obtenues. Celles-ci sont identiques les unes aux autres, à partir des banques
d'ADNc qui ont été amplifiées. Celles-ci pourraient être un véritable reflet
des espèces d'ARN abondantes, ou ils pourraient être des artefacts de PCR. Une
façon d’identifier l’origine de ces lectures courtes est de déterminer si les
mêmes séquences sont observées dans différentes répliques biologiques.
Une autre considération importante concernant
la construction de la bibliothèque est de savoir s'il faut ou non préparer des
bibliothèques spécifiques aux brins. Ces bibliothèques ont l'avantage de
fournir des informations sur l'orientation des transcrits, ce qui est très
utile pour l'annotation du transcriptome, en particulier pour les régions ayant
des transcriptions chevauchantes et de directions opposées. Cependant, les bibliothèques
spécifiques aux brins sont laborieuses à produire parce qu'elles nécessitent de
nombreuses étapes ou une ligation ARN-ARN directe, qui est peu efficace. En
outre, il est essentiel de s'assurer que les transcrits anti-sens ne sont pas
des artefacts de transcription inverse. En raison de ces complications, la
plupart des études jusqu'à présent ont analysé des ADNc sans information de
brin. (9)
2. ROGLER, C. E., TCHAIKOVSKAYA, Tatyana,
NOREL, Raquel, MASSIMI, Aldo, PLESCIA, Christopher, RUBASHEVSKY, Eugeny,
SIEBERT, Paul et ROGLER, Leslie E. RNA expression microarrays (REMs), a
high-throughput method to measure differences in gene expression in diverse
biological samples. Nucleic Acids Research. 2004. Vol. 32,
n° 15, pp. e120. DOI 10.1093/nar/gnh116. PMID: 15329382PMCID:
PMC516075
9. WANG,
Zhong, GERSTEIN, Mark et SNYDER, Michael. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews. Genetics. janvier 2009.
Vol. 10, n° 1, pp. 57‑63. DOI 10.1038/nrg2484. PMID:
19015660PMCID: PMC2949280