Wiki des méthodes de biologie moléculaire et cellulaire
Glossaire collaboratif des méthodes expérimentales de biologie moléculaire et cellulaire. Ce glossaire est réalisé par les étudiants du module "Méthodes expérimentales de biologie moléculaire et cellulaire" sous la supervision de l'équipe de Génétique Animale.
Spécial | A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z | Tout
H |
|---|
| JG | HCR RNA FISH | |
|---|---|---|
HCR RNA FISH I) Objectifs La méthode HCR RNA FISH (Hybridization Chain Reaction RNA Fluorescence In Situ Hybridization) est une technique de détection d’ARN in situ basée sur l’hybridation et l'amplification de sondes fluorescentes. Elle est caractérisée par une grande spécificité grâce à l’utilisation de sondes spécifiques et permet même de réaliser du multi-ciblage grâce à l'utilisation de différentes couleurs de fluorophores (Huang et al., 2023). De plus, la fluorescence des fluorophores présents sur les sondes est visible seulement lorsque les sondes sont hybridées, permettant ainsi la réduction des signaux parasites. Cette méthode est utilisée pour l’étude de l’expression de gènes afin d’identifier où et quand est exprimé un gène. Elle peut donc être utilisée dans de très nombreux domaines comme les neurosciences, la cancérologie, la microbiologie ou encore la biologie du développement. II) Principe Cette technique utilise deux types de sondes : des sondes initiatrices et des sondes d’amplification. Les sondes initiatrices sont designées pour se fixer spécifiquement à l’ARN cible. Une fois hybridées avec l’ARN, débute la phase d’amplification du signal : leur extrémité libre peut s’apparier avec les sondes d’amplification, qui s’assemblent ensuite en chaîne par hybridation successive. Ces sondes d’amplification h1 et h2, initialement en épingle et préalablement marquées par des fluorophores, sont designées telles que l’extrémité 3’ de h1 soit complémentaire de l’extrémité 5’ de h2 et réciproquement, ce qui permet leur hybridation en chaîne. La fluorescence est activée par l’hybridation des sondes. La détection des cellules ou tissus dans lesquels l’ARN d'intérêt est produit est alors possible à l’aide d’un microscope à fluorescence.
Figure 1. : (from Choi et al., 2018). In situ HCR v3.0 using split-initiator probes. (A) HCR mechanism. Green stars denote fluorophores. Arrowhead indicates 3′ end of each strand. (B) Standard probes carry full HCR initiator I1 and generate amplified background if they bind non-specifically. Split-initiator probes P1 and P2 each carry half of HCR initiator I1 and do not generate amplified background if they bind non-specifically. (C) Two-stage in situ HCR protocol. Detection stage : probe sets hybridize to mRNA targets, unused probes are washed from the sample. Amplification stage: specifically bound probe pairs trigger self-assembly of a tethered fluorescent amplification polymer and unused hairpins are washed from the sample. Automatic background suppression throughout the protocol: any reagents that bind non-specifically do not lead to generation of amplified background. (D) Multiplexing timeline. The same two-stage protocol is used independent of the number of target mRNAs. HCR amplification is performed overnight for qHCR imaging and qHCR flow cytometry experiments (to maximize the signal-to-background ratio) and for 45-90 min for dHCR imaging experiments (to resolve individual molecules as diffraction-limited dots). III) Mode opératoire L’échantillon est d’abord fixé pour préserver sa morphologie et ses composants biologiques, puis perméabilisé pour faciliter la diffusion des réactifs. Après une étape de pré-hybridation pour bloquer les sites non spécifiques, les sondes HCR™ HiFi complémentaires à l’ARN cible sont hybridées. Les sondes non fixées sont ensuite éliminées par lavage. L’échantillon est préparé pour l’amplification avec un tampon spécifique, puis les amplificateurs HCR™ Gold (hairpins h1 et h2) sont ajoutés pour générer un signal fluorescent. Un second lavage élimine les amplificateurs non liés. Une contre-coloration optionnelle (DAPI, Hoechst) permet de marquer les noyaux. L’échantillon est enfin monté entre lame et lamelle et imagé par microscopie à fluorescence (Molecular Instruments, 2025). IV) Présentation des résultats Les résultats sont des images de microscopie à fluorescence.
Figure 2 : Image au microscope à fluorescence d’un embryon de Drosophila, avec la région antérieure à gauche et postérieure à droite. Les ARNm du gène shavenbaby (svb) sont marqués par HCR RNA FISH en magenta et les noyaux des cellules sont colorés en blanc par DAPI. V) Interprétation des résultats Cette méthode permet à la fois une analyse qualitative, avec localisation des cellules où l'ARN cible est exprimé, et une analyse quantitative car l'intensité de la fluorescence est directement liée à la quantité d’ARN cibles. VI) Intérêts et limites Les intérêts de cette technique sont une visualisation directe des ARN in situ avec un grande spécificité ainsi que la localisation et quantification de plusieurs ARN simultanément (Huang et al., 2023). De plus, par rapport à une méthode RNA FISH classique, elle a l’avantage de ne pas utiliser d’anticorps. Les limites de cette technique sont les mêmes qu’une méthode RNA FISH classique (cf wiki FISH (Fluorescence In Situ Hybridization)). La fluorescence est sensible à la lumière et limitée dans le temps car son intensité diminue avec le temps. L’observation doit donc être faite rapidement après le montage. Cette technique reste chère, car il faut se procurer les sondes spécifiques aux régions à tester. Enfin, cette méthode est difficile à appliquer in vivo. VII) Références bibliographiques Choi HMT, Schwarzkopf M, Fornace ME, Acharya A, Artavanis G, Stegmaier J, Cunha A, Pierce NA. (2018). Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145 (dev165753). Huang T, Guillotin B, Rahni R, Birnbaum KD, Wagner D. (2023). A rapid and sensitive, multiplex, whole mount RNA fluorescence in situ hybridization and immunohistochemistry protocol. Plant Methods 19, 131. Molecular Instruments. (2025). HCRTM Gold RNA-FISH | Molecular Instruments, Inc. HCRTM Gold RNA-FISH, https://www.molecularinstruments.com/hcr-gold-rnafish
| ||


| OD | HPLC | |
|---|---|---|
High Performance Liquid Chromatography (HPLC) I) Objectifs L’objectif de la chromatographie en phase liquide est de séparer les différentes molécules d’un mélange liquide afin de connaître sa composition ou dans un but préparatif, c’est à dire de purification de molécules. II) Principe A- Principe général Le principe de la HPLC est, comme pour les autres variantes de chromatographie, d’utiliser les différences de propriétés physico-chimiques de différents composés pour les séparer. Un liquide, l’éluant, constitue la phase mobile, qui va entraîner plus ou moins facilement les molécules du mélange. Une phase dite stationnaire va permettre de séparer les solutés en interagissant avec eux. Cette phase stationnaire est composée d’un support de porosité variable recouvert d’un gel spécifique choisi en fonction des molécules à séparer. Le support se trouve sous forme de micro-billes maintenues dans la colonne. B- Les différentes variantes de HPLC L’utilisation de la HPLC par absorption est réservée à la séparation de molécules en fonction de leur polarité et de leur hydrophobie. Les solutés sont absorbés de manière différentielle par la phase stationnaire, selon l’intensité des liaisons faibles formées et leurs solubilités dans l’éluant. La résultante de ces deux forces (rétention et entraînement) cause la migration différentielle des composés qui peuvent ensuite être séparés. Le principe de la HPLC de partage en phase inverse repose sur des greffes composées de longues molécules fixées sur le support par des liaisons covalentes. La phase inverse signifie que le solvant est plus polaire que la phase stationnaire, et les solutés sont séparés en fonction de leur hydrophobie. La greffe peut être par exemple des radicaux alkyles (chaînes C18). Selon les affinités des solutés pour la greffe, ceux-ci vont être entraîné par la phase mobile [1]. Il est possible de réaliser un gradient de force éluante en changeant les éluants utilisés pour pouvoir séparer des molécules très différentes. La HPLC par échange d’ions vise à séparer des ions en se basant sur leur différence d’affinité pour une greffe ionique [2]. Ainsi, les ions du mélange vont se substituer aux ions de la greffe, puis après un changement d’éluant contenant un troisième type d’ions, vont être remplacés par ceux-ci. Ils vont donc migrer plus ou moins facilement et ainsi être séparés. La HPLC par exclusion stérique vise à trier les molécules en fonction de leur taille. La colonne contient des billes de silices poreuses qui vont retenir les plus petites molécules et laisser passer les plus grosses. Cette technique est un bon moyen de récupérer les grosses molécules mais ne permet pas de purifier les petites qui restent coincées dans les pores des billes de silice [1]. La HPLC d’affinité est basé sur des interactions de type ligand-enzyme ou anticorps-antigène. Cette méthode est très utile pour séparer les protéines. Les molécules sont séparées en fonction de leur affinité pour le ligand greffé à la silice de la colonne [1].
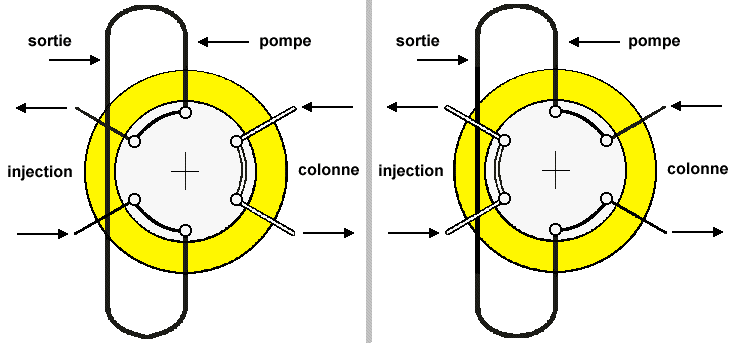
III) Mode opératoire L’échantillon liquide à analyser est placé dans un injecteur et envoyé dans la boucle d’injection. Après avoir été dégazé, l’éluant est pompé à son tour dans la boucle, grâce à des pompes constituées de saphir industriel (inerte et très dur), ce qui permet de délivrer un débit précis, constant voire élevé [1]. Ces pompes permettent de travailler en mode isocratique, c’est à dire avec un mélange d’éluants de composition constante, ou en mode gradient, autrement dit avec une variation linéaire de la composition du mélange d’éluants, ou par palier, si on le souhaite.
Figure 1: Schéma d’une boucle d’injection en phase d’injection et de balayage [5] La boucle d’injection effectue une rotation entre les phases d’injection et de balayage pour un vidage plus simple de la colonne Le mélange passe ensuite dans la colonne en acier inoxydable de quelques dizaines de centimètres de long pouvant supporter de fortes pressions. Elle est constituée de microbilles de silice greffées de molécules qui varient en fonction du type d’échantillon à analyser [1]. A la sortie de la colonne, les composés entrent en contact avec le détecteur à des temps différents. Le détecteur le plus utilisé en HPLC est un spectrophotomètre. Il est constitué d'une cuve à circulation en quartz, d'une capacité d'environ 10 µl, traversée en continu par un faisceau U.V. D’autres détecteurs utilisant la spectrométrie de masse, la fluorescence, ou l’indice de réfraction peuvent être utilisés [2]. Le signal généré par le détecteur est transmis au calculateur-enregistreur, qui va analyser et convertir les résultats pour les afficher sous la forme d’un chromatogramme. Bien que le mode opératoire reste globalement le même, des catalogues répertorient les différents paramètres en fonction de la machine utilisée et de la nature des molécules de l’échantillon.
IV) Présentation des résultats
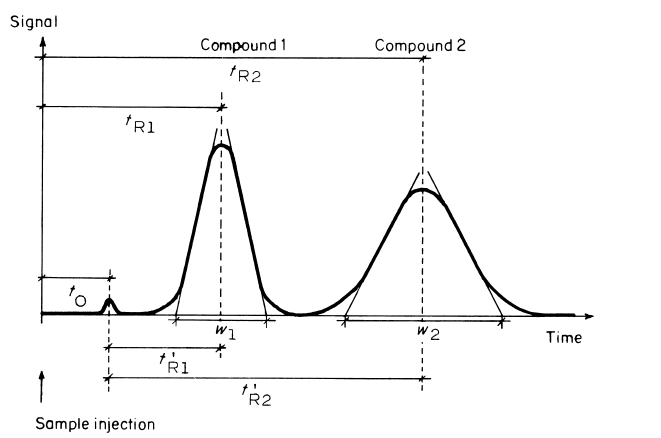
Le résultat d’une HPLC est présenté sous forme d’un chromatogramme. L’abscisse correspond au temps de rétention, et donc à la facilité avec laquelle la molécule a traversé la colonne. C’est une constante pour une condition de chromatographie donnée. L’ordonnée correspond quant à elle à l’intensité du signal, c’est à dire à la quantité de la molécule détectée au temps t. On peut donc en déduire la quantité totale de la molécule dans le mélange en calculant l’aire du pic correspondant. Enfin, t0 correspond au temps de rétention d’une molécule qui n’est pas retenue par la phase stationnaire.
Figure 2: Les différentes grandeurs d’un chromatogramme [1] Abscisse : temps de rétention en minutes, ordonnée : intensité du signal ; tr1 et tr2 sont les temps de rétention des composés respectifs, t0 est le temps mort, c’est à dire le temps de rétention d’un composé non retenu par la colonne) et ω1 et ω2 sont les largeurs des pics à leurs bases.
V) Interprétation des résultats Chaque pic traduit la présence d’une molécule dans l’échantillon. Il est possible de quantifier les molécules en mesurant l’aire des pics. Il existe plusieurs méthodes pour approximer cette aire : soit par triangulation (figure 2), soit en reportant la valeur calculée automatiquement par la machine ou le logiciel. La proportion de chaque composé est déterminée par le rapport entre l’aire et la somme de toute les aires. Le facteur de résolution permet de déterminer la significativité des résultats, il mesure la séparation des pics et permet d’évaluer la présence de chevauchement. Il vaut [1] :
où tr1 et tr2 sont les temps de rétention des composés respectifs, et ω1 et ω2 sont les largeurs des pics à leurs bases.
V I ) Intérêts et limites La HPLC est une technique haute résolution et simple à mettre en œuvre, qui permet d’identifier et de quantifier de manière très précise les composants d’un mélange. Elle permet d’analyser tous les types de molécules tant qu’elles sont en phase liquide, et n’est pas limitée aux molécules volatiles comme la chromatographie en phase gazeuse. Elle permet aussi de purifier les produits en récupérant dans des tubes différents des composants en fonction du temps. Cependant, cette méthode peut être onéreuse pour certains laboratoires. Le prix par échantillon est important (de l’ordre de 200 €) et est dû à la quantité d’éluant utilisé qui peut être de plusieurs litres. En effet, le débit de la phase mobile est de l’ordre de 5 ml/min, et la durée de l’analyse de l’ordre de l’heure [3]. 100 ml d’acétonitrile coûtent plus de 60€ et 100 ml de méthanol coûtent 50€ [4]. De plus, le prix des machines est conséquent à l’achat (40 000-50 000 € pour les plus simples) et à l'entretien. Cette technique est surtout consacrée aux molécules organiques solubles dans un liquide ou ioniques.
VII) Références bibliographiques
[1] CUQ J.-L. (2007). Cours sur la Chromatographie liquide. Université de Montpellier 2 science et technique. [2] Meyer V.R. (2004). Practical High-Performance Liquid Chromatography Fourth edition. ISBN: 0-470-09377-3 [3] http://www.doping.chuv.ch/files/presentation-lc-fr.pdf [4] http://www.sigmaaldrich.com/france.html [5] http://www.rocler.qc.ca/pdubreui/chromatographie/HPLC/chroma3.html
Nous remercions Daniel CATHELINE, ingénieur de recherche au laboratoire de biochimie et nutrition humaine d’AGROCAMPUS OUEST pour le temps qu’il nous a consacré ainsi que pour ses explications sur la HPLC.
| ||
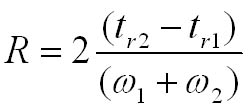
| NK | Hybridation génomique comparative | |
|---|---|---|
Hybridation génomique comparative.
I) Objectifs Les anomalies chromosomiques désignent l’aneuploïdie avec la perte ou l’ajout de chromosomes entiers, mais aussi d’autres réarrangements chromosomiques comme la délétion, la duplication ou la translocation de morceaux de chromosomes (Béri-Dexheimer, 2005). Ces anomalies entraînent des conséquences variées tels que les retards de développement, l’autisme et d’autres anomalies congénitales (Shinawi et Cheung, 2008). L’hybridation génomique comparative (HGC) est une méthode utilisée pour la détection de ces pathologies, notamment au stade fœtal (Jonveaux, 2010), et se révèle être prometteuse dans la recherche sur le cancer. II) Principe L’hybridation génomique comparative se base sur les déséquilibres génomiques pour diagnostiquer ces anomalies chromosomiques. La technique est actuellement utilisée sur puce à ADN pour plus d’efficacité et de précision, diminuant ainsi la taille des déséquilibres détectables. La technique repose sur l’hybridation d’ADN de référence et d’ADN à tester (Shinawi, et Cheung, 2008). III) Mode opératoire (Figure 1) La puce d’hybridation génomique comparative est recouverte de séquences d’ADN cibles de longueur variable en fonction de la précision voulue. Deux sortes de sondes sont préparées : l’ADN à tester marqué par la Cyanine 3 et l’ADN de référence marqué par la Cyanine 5. De l’ADN Cot-1 est ajouté pour éviter l'hybridation de séquences répétées. Les deux sondes sont ensuite déposées sur la lame pour l’hybridation. La puce est ensuite lavée et lue à l’aide d’un scanner laser. Les intensités de fluorescence des ADN marqués à la Cyanine 3 et à la Cyanine 5 sont comparées à l’aide d’une représentation graphique (Béri-Dexheimer et al, 2007).
Figure 1 : Principe de l'hybridation génomique comparative sur microréseau d'ADN (Béri-Dexheimer et al, 2007). IV) Présentation des résultats Après hybridation, la fluorescence est capturée à l’aide d’une caméra (HGC métaphasique) ou d’un scanner laser pour l’hybridation génomique comparative sur microréseau cette image montre déjà des premiers résultats. En effet, les points rouges indiquent une perte de l’ADN test, les points verts un gain de l’ADN test et les points jaunes indiquent la présence d’une quantité équivalente entre l’ADN test et l’ADN témoin. Pour une analyse quantitative, un logiciel calcule les rapports de fluorescence entre les deux ADN, et représente les résultats sous forme de graphique (Jonveaux, 2010) (Figure 2). L’analyse est réalisée pour l’ensemble des cibles présentes sur la lame. Chaque point du graphique correspond à une cible dont la cartographie physique sur le chromosome est connue permettant ainsi de quantifier la taille de l’anomalie et d’apprécier les gènes impliqués dans le remaniement (Béri-Dexheimer et al, 2007).
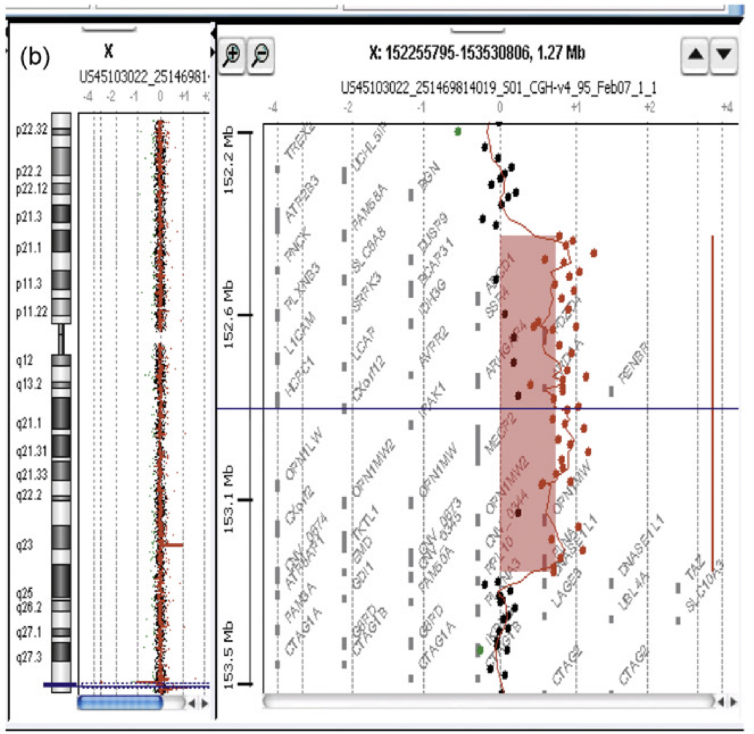
Figure 2 : Exemple d’un rapport d’une puce à oligonucléotides de la firme Agilent Technologies avec identification d’un gain en Xq28 (Jonveaux, 2010). À gauche est représenté le pictogramme du chromosome X avec une déviation reconnue par le logiciel dans la partie terminale du bras long du chromosome X (ligne bleue). À droite est représenté un zoom de la région en déséquilibre avec chaque point rouge du graphique correspondant à une cible. V) Interprétation des résultats L’interprétation quantitative des résultats de l’hybridation génomique comparative se fait par l’interprétation de la courbe de résultats (figure 2). En pratique, pour chaque cible étudiée,
Pour des problèmes de représentation graphique, la plupart des auteurs utilisent le log 2 de ce ratio. Sur la figure 1, la position génomique de chaque cible est donnée en Mb par rapport au télomère du chromosome étudié. De plus, l’échelle des ratios a été modifiée et une déviation à +1 correspond à un gain, tandis qu’une déviation à -1 correspond à une perte. Ici, il s’agit d’un gain d’une taille de 751 Kb s’étendant sur la séquence de référence du génome humain des nucléotides 152 511 063 à 153 262 357. VI) Intérêts et limites La méthode d’hybridation génomique comparative a de nombreuses fois montré son efficacité, cependant, elle possède quelques limites. Premièrement, son incapacité à détecter les réarrangements de structure équilibrés, ce qui peut conduire à des erreurs dans l'identification des anomalies génétiques. De plus, cette méthode ne permet pas la détection des anomalies causées par des interruptions de gènes. Elle cause aussi une limitation dans la détection des anomalies de la ploïdie telles que la triploïdie, qui ne sont pas identifiables car les échantillons test et témoin contiennent la même proportion de lot haploïde par quantité d'ADN hybridée. Enfin, avec cette méthode, l’interprétation des variations numériques chromosomiques est complexe, et seule une collecte extensive de données cliniques et biologiques sur de grandes populations peut aider à clarifier le caractère pathogène de certaines de ces variations. VII) Références bibliographiques Béri-Dexheimer M. (2005). Hybridation génomique comparative en microréseau : évolutions techniques et place dans la stratégie diagnostique, expérience dans le cadre du retard mental. Sciences pharmaceutiques. Béri-Dexheimer M., Bonnet C., Chambon P., Brochet K., Grégoire M.-J., Jonveaux P. (2007). L'hybridation génomique comparative sur microréseau d'ADN (puces à ADN) en pathologie chromosomique constitutionnelle, Pathologie Biologie, Volume 55, Issue 1, p.13-18. Jonveaux P. (2010). Technique d’hybridation génomique comparative sur microréseau d’ADN et fœtopathologie, Archives de Pédiatrie, Volume 17, Issue 7, p.1119-1123. Shinawi M., Cheung S. W. (2008). The array CGH and its clinical applications, Drug Discovery Today, Volume 13, Issues 17–18, p.760-770.
| ||