Wiki des méthodes de biologie moléculaire et cellulaire
Glossaire collaboratif des méthodes expérimentales de biologie moléculaire et cellulaire. Ce glossaire est réalisé par les étudiants du module "Méthodes expérimentales de biologie moléculaire et cellulaire" sous la supervision de l'équipe de Génétique Animale.
Spécial | A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z | Tout
E |
|---|
| SC | Electrophorèse Capillaire sur Gel (CGE) | ||
|---|---|---|---|
Electrophorèse Capillaire sur Gel (CGE)
I) Objectifs L’électrophorèse capillaire sur gel (CGE) a été décrite par Hjertén en 19831 et permet la séparation des molécules en fonction de leur taille et de leur charge. Cette méthode séparative est utilisée pour identifier et quantifier des molécules (telles que des protéines et acides nucléiques) soumises à un champ électrique et passant dans un capillaire de faible diamètre en silice. Elle peut également être utilisée pour mesurer la pureté d’une solution. | |||
| NM | EMSA – Le retard sur gel (Electrophoretic mobility shift assay) | |
|---|---|---|
EMSA – Le retard sur gel (Electrophoretic mobility shift assay)
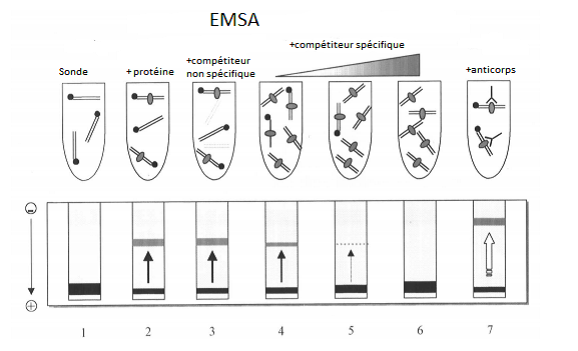
I. ObjectifsLe retard sur gel (EMSA), décrit pour la première fois par Garner et Revzin, et Fried et Crothers en 1981, est utilisé pour détecter in vitro une interaction entre l’ADN et des protéines (facteurs de transcription). II. PrincipeLa régulation de la transcription est réalisée par plusieurs protéines, liées directement ou indirectement aux régions régulatrices de l’ADN. Ces protéines sont appelées facteurs de transcription. Leur liaison aux promoteurs des gènes permet d’activer ou de réprimer leur expression. La méthode plus utilisée pour étudier les liaisons « ADN–protéine » est l’EMSA. Elle est basée sur le fait que la mobilité électrophorétique du complexe « ADN–protéine » est différente de la mobilité de l’ADN « libre », générant donc une différence de taille entre les molécules libres et les complexes. Lors de l’électrophorèse, le mélange ADN–protéine est retardé (décalé = shift) sur le gel, par rapport à l’ADN seul. III. Mode opératoireL’expérience est réalisée avec des sondes d’ADN, marquées au phosphore radioactif (32P), et incubées avec la protéine dans un tampon approprié. Ce mélange est soumis à électrophorèse en gel de polyacrylamide en conditions non dénaturantes. Les interactions détectées entre l’ADN et la protéine par l’EMSA ne sont pas nécessairement spécifiques. II faut donc faire des essais de compétition pour différencier la liaison spécifique de la liaison non spécifique. Deux sondes non radioactives sont utilisées : une des sondes est une séquence anonyme (compétiteur non spécifique) et l’autre sonde est la sonde originale non marquée (compétiteur spécifique ou sonde froide). Elle est ajoutée au mélange en quantités supérieures et croissantes par rapport à la sonde marquée (sonde chaude). L’identité de la protéine qui forme le complexe peut être révélée grâce à des anticorps spécifiques dirigés contre les facteurs protéiques « suspects ». IV. Présentation des résultatsSi la protéine se fixe effectivement sur le fragment d’ADN, la migration électrophorétique de ce dernier sera plus lente, et la bande correspondante migrera moins loin sur le gel que la bande correspondant à l’ADN pur. V. Interprétation des résultatsSi l’interaction initiale entre la sonde marquée (chaude) et la protéine est spécifique, le compétiteur spécifique (froid) se liera au complexe et le bande lourde sur le gel s’estompera, puis disparaitra (Figure 1). L’ajout d’un compétiteur non spécifique ne change pas l’apparence du complexe original, même en grandes concentrations. L’ajout d’un anticorps spécifique de la protéine conduit à la formation d’un super complexe (supershift) dont la mobilité est encore plus faible, permettant de confirmer l’identité du facteur suspect. Figure 1 : Interprétation du résultat après l’électrophorèse (Lin et Barbosa, 2002)
VI. Intérêts et limitesAvec certaines adaptations, la méthode peut aussi mesurer des paramètres cinétiques et évaluer la spécificité de l’interaction entre l’ADN et la protéine. Mutations et délétions peuvent être introduites dans les sondes pour vérifier quelles sont les bases importantes pour la liaison entre l’ADN et la protéine. Pour ces raisons, la méthode est de plus appliquée dans l’étude des interactions entre les éléments cis et trans. Par contre, la méthode est très longue, laborieuse, et il faut avoir une grande quantité d’échantillon. Le principal inconvénient est la nécessité de manipuler des matériaux radioactifs Il faut avoir une formation spécifique, et être prudent avec le devenir des déchets et le stockage des sondes. Pour réduire le temps, la quantité d’échantillon nécessaire et éviter le marquage isotopique, diverses méthodes ont été développées, utilisant la technique d’électrophorèse capillaire. Le CEMSA a permis d’éliminer plusieurs inconvénients de la méthode classique et il a l’avantage d’utiliser la fluorescence, et de permettre la détermination des constantes de liaison. Mais, les instruments nécessaires au CEMSA ne sont pas disponibles dans les tous laboratoires et il faut avoir une grande connaissance technique.
VII. Références bibliographiquesFried M, Crothers DM. Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res 1981;9(23):6505-25. Garner MM, Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res 1981;9(13):3047-60. HILLEBRAND, Sandro. Estudos de Reconhecimento biomolecular por eletroforese capilar. 2005. 81 f. Tese (Doutorado) - Curso de Ciências, Universidade de São Paulo, São Carlos, 2005. Jin CJ, Barbosa AS. Técnicas de Análise da Regulação da Transcrição Gênica e suas Aplicações na Endocrinologia Molecular. Arq Bras Endocrinol Metab vol 46 nº 4 Agosto 2002 | ||
| VC | EXTRACTION DES LIPIDES TOTAUX ET TRANSFORMATION EN ESTER METHYLIQUE EN VUE D’UNE ANALYSE D’ACIDE GRAS EN CHROMATOGRAPHIE GAZEUSE | |
|---|---|---|
Extraction des lipides totaux et transformation en ester méthylique en vue d’une analyse d’acide gras en chromatographie gazeuse I) I) Objectifs L’objectif de cette méthode est de permettre l’extraction et la transformation des lipides totaux d’un échantillon afin de réaliser une analyse d’acide gras en chromatographie en phase gazeuse. A partir d’un échantillon biologique quelconque (tissu, lysat cellulaire, …), cette méthode permet la séparation et l’extraction des lipides totaux et leur transformation en ester méthylique, afin d’analyser qualitativement et quantitativement la nature des constituants. Cette analyse se base sur l’utilisation d’un étalon interne. II) II) Principe Les lipides à extraire de l’échantillon doivent être séparés des autres constituants comme les glucides ou les protéines. Pour une séparation simple et rapide, on utilise une des propriétés physicochimiques des lipides : leur hydrophobie. Grâce à l’utilisation de solvants organiques tels que l’hexane, l’isopropanol ou le pentane, mis en contact avec l’échantillon, les lipides vont être extraits des autres constituants par la formation de deux phases. La phase organique, ici, la phase supérieure, contiendra les lipides totaux et la phase aqueuse contiendra le reste des constituants. Une fois les lipides isolés du mélange, des réactions chimiques de saponification, puis de méthylation vont être réalisées afin d’obtenir des esters méthyliques, qui sont facilement analysables en chromatographie gazeuse. Afin de pouvoir quantifier la nature des acides gras de l’échantillon, l’utilisation d’un standard interne absent de l’échantillon est nécessaire. Le standard couramment utilisé est un acide gras saturé impair : l’acide heptadécanoïque (C17:0) ou l’acide tridécanoïque (C13:0). III) III) Mode opératoire L’échantillon en solution (lysat cellulaire, broyat de tissu solubilisé, …) est ajouté dans un tube à méthylation à bouchon fermé. La première étape est l’ajout d’une quantité connue et précise du standard interne. La quantité de standard à ajouter est fonction de la quantité estimée de lipides dans l’échantillon. Pour une quantification plus précise, il est nécessaire d’avoir une quantité de standard interne qui est du même ordre de grandeur que les acides gras majoritaires présents dans l’échantillon. Une acidification de la solution est réalisée en ajoutant 1 mL de solution d’HCL 3M.1 Puis, 4 mL d’un mélange hexane / isopropanol (3:2 v/v) sont ajoutés. Une homogénéisation est nécessaire afin de bien séparer les constituants dans les phases. La solution est ensuite centrifugée (1100 g, 10 min) et la phase supérieure organique est prélevée et transférée dans un nouveau tube de méthylation. Cette étape d’extraction est répétée sur la phase aqueuse avec 2 mL d’hexane / isopropanol pour récupérer les lipides résiduels n’ayant pas été extraits. Un lavage est effectué en ajoutant 2 mL de solution NaCl (0,9 %, v/v) puis on centrifuge (3 min, 1100 g) et la phase organique est récoltée. Une évaporation pour éliminer totalement les solvants est effectuée (sous flux d’azote, ou sous vide). Ensuite, une étape de saponification est réalisée. 1 mL de NaOH 0,5M est ajouté avant la mise au bain-marie à 70°C pendant 20 min des échantillons. L’étape de méthylation passe par l’ajout d’1 mL de Boron trifluoride dans le méthanol (12 %, v/v). Une double extraction est de nouveau réalisée en utilisant 4 mL de NaCl (0,9 %, v/v) additionné de 4 mL de pentane. La phase organique est ensuite lavée avec 2 mL de NaCl (0,9%, v/v) et évaporée. Les esters méthyliques obtenus sont solubilisés dans 150 µL d’hexane et peuvent être analysés en chromatographie gazeuse. IV) IV) Présentation des résultats Les esters méthyliques sont analysés en chromatographie gazeuse, couplée à un spectrophotomètre de masse par exemple2. On obtient un chromatogramme avec lequel on peut identifier et quantifier les acides gras de l’échantillon.
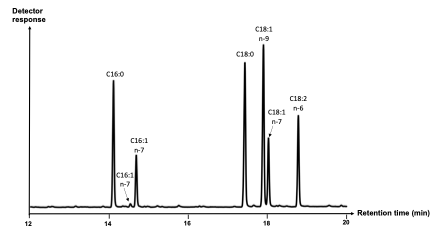
Figure 1 : Chromatogramme d’analyse de certains acides gras d’hépatocarninome humain, obtenu par chromatographie gazeuse. Chaque pic correspond à un acide gras, identifiés avec le spectre de masse (production personnelle).
V) V) Interprétation des résultats Les acides gras présents dans l’échantillon sont identifiés grâce à leur temps de rétention et leur spectre de masse. Une comparaison avec des molécules de références pour les identifier est possible grâce à l’utilisation d’une banque. Pour certains acides gras polyinsaturés peu communs, il est parfois nécessaire d’effectuer des dérivations pour identifier spécifiquement la position des insaturations3, 4. VI) VI) Intérêts et limites Cette extraction de lipides suivie d’une préparation d’analyse en chromatographie gazeuse par transformation en ester méthylique permet une analyse fine des constituants en acides gras de l’échantillon. De plus, elle est qualitative et quantitative via l’utilisation d’un standard. Cependant elle ne permet pas d’identifier spécifiquement les classes des lipides totaux (phospholipides, triglycérides, esters de cholestérol, acides gras non estérifiés). Pour cela, une séparation des lipides totaux en fonction des classes est nécessaire par séparation en chromatographie sur couche mince. VII) VII) Références bibliographiques 1. Rioux, V., F. Pédrono, H. Blanchard, C. Duby, N. Boulier-Monthéan, L. Bernard, E. Beauchamp, D. Catheline, and P. Legrand (2013) “Trans-Vaccenate Is Δ13-Desaturated by FADS3 in Rodents.” Journal of Lipid Research 54 (12): 3438–52. 2. Rioux, V., B. Choque, H. Ezanno, C. Duby, D. Catheline, and P. Legrand (2015) “Influence of the Cis-9, Cis-12 and Cis-15 Double Bond Position in Octadecenoic Acid (18:1) Isomers on the Rat FADS2-Catalyzed Δ6-Desaturation.” Chemistry and Physics of Lipids 187 (April): 10–19. 3. Guillou, H., V. Rioux, D. Catheline, J. N. Thibault, M. Bouriel, S. Jan, S. D’Andrea, and P. Legrand (2003) “Conversion of hexade- canoic acid to hexadecenoic acid by rat Delta 6-desaturase”. J. Lipid Res. 44: 450–454. 4. Christie, W. W., G. Dobson, and R. O. Adlof (2007). “A practical guide to the isolation, analysis and identification of conjugated linoleic acid. Lipids. 42: 1073–1084.
| ||

| AP | Extraction et purification d'ARN par le réactif TRIZOL® : Amélioration de la méthode Chomczynski et Sacchi | |
|---|---|---|
I) Objectifs L’extraction de l’ARN au thiocyanate de guanidine a été mise en évidence en 1987 par les biochimistes Piotr Chomczynski et Nicoletta Sacchi [2], puis remise à jour en 2006 par ces mêmes chercheurs [3]. Cette technique est un procédé visant à isoler les ARN totaux d’un échantillon biologique, permettant par la suite l’étude de l’expression des gènes. II) Principe Les ARN totaux sont isolés par la réalisation d’une unique extraction entraînant une acidification du pH de la solution. Ceci permet un isolement des ARN dans une phase aqueuse supérieure, tandis que les ADN et protéines, moins solubles, se retrouvent dans l’interphase ou dans la phase organique inférieure [3]. Cette technique s’est imposée comme méthode de référence et est idéale pour une extraction rapide de plusieurs échantillons simultanément. Afin d’apprécier la qualité des ARN obtenus, il est possible d'effectuer un dosage spectrophotométrique ainsi qu’une électrophorèse. Etant donné la fiabilité de la méthode, l’obtention d’échantillons purs est attendue. Les ARN totaux obtenus peuvent être utilisés dans le cadre de divers méthodes tel qu’une RT-PCR, un Northern Blot ou encore une hybridation. III) Mode opératoire La première étape consiste en la lyse des cellules ou tissus d’intérêts par le TRizol® constitué de thiocyanate de guanidine (agent dénaturant des protéines) et de phénol (solvant organique). Après homogénéisation des produits de la lyse, les ARN totaux sont purifiés par du chloroforme. Après centrifugation, ils se retrouvent alors dans la phase aqueuse supérieure (Figure 1). Une précipitation de ces ARN par de l’isopropanol est ensuite réalisée. En fin d’opération, les ARN sont souvent contaminés par le phénol. Ils peuvent être purifiés par une deuxième précipitation par de l’éthanol, qui devra lui-même être totalement éliminé, afin d’éviter des problèmes de bruits de fond. Enfin, l’ADN persistant est traité par de la DNase [6]. On obtient en fin de méthode des ARN totaux purifiés, exempts de tout solvant organique. Afin d’évaluer la quantité et le niveau de pureté des ARN extraits, des dosages spectrophotométriques à 230, 260 nm et 280 nm peuvent être réalisés, alors que la qualité de ces ARN est estimée par une électrophorèse [3].
Figure 1 : Observation des phases organique et aqueuse après extraction des ARN [5]. IV) Présentation des résultats Les principaux résultats de cette méthode d'extraction sont donc des résultats de qualité, de quantité et de pureté, primordiaux pour assurer l’utilisation correcte des ARN par la suite (Figure 2).
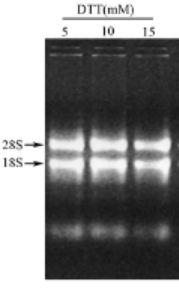
Figure 2 : Electrophorèse obtenue après l'extraction d’ARN de siliques d’Arabidopsis au TRIzol® en présence de quantité variable de DTT [4].
V) Interprétation des résultats
Les ARN sont considérés comme purs si, lors du dosage spectrophotométrique, les 2 rapports d’absorbance (A260 / A230) et (A260 / A280) sont proches de 2. Ces résultats seront confirmés par la réalisation d’une électrophorèse (Figure 2). Si l’on observe trois fortes bandes correspondant aux ARN ribosomiques 28S, 18S et 5S alors les ARN extraits sont purs. VI) Intérêts et limites Comparée aux autres méthodes d'extraction existantes, comme par exemple la méthode Chirgwin et al. [1] ou la méthode d’extraction sur colonne [6], l’extraction au phénol-chloroforme peut être caractérisée par sa fiabilité en termes de qualité des ARN totaux obtenus [4]. De plus, cette méthode est simple et rapide. En effet, elle nécessite une unique extraction et permet l’isolement simultané de plusieurs échantillons. Au contraire, l’ancienne méthode fortement utilisée, la méthode Chirgwin et al. [1], requiert de nombreuses ultracentrifugations et est plus longue [3]. Cependant, la méthode d’extraction au thiocyanate de guanidine reste plus lente que la méthode d’extraction sur colonne [6]. Références bibliographiques [1] Chirgwin J. Przybyla A, MacDonald R., Rutter W. (1979). Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 18 (24). 5294-5299.
[2] Chomczynski P., Sacchi N. (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 162. 156-9.7
[3] Chomczynski P., Sacchi N. (2006). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: Twenty-something years on. Nature protocols. 1. 581-5.
[4] Meng L., Feldman L. (2010). A rapid TRIzol-based two-step method for DNA-free RNA extraction from Arabidopsis siliques and dry seeds. Biotechnology journal. 5. 183-186. https://openwetware.org/wiki/RNA_extraction_using_trizol/tri. Consulté le 13/03/2020.
[6] Tolosa J., Schjenken J., Civiti T., Clifton V., Smith R. (2007). Column-based method to simultaneously extract DNA, RNA, and proteins from the same sample. BioTechniques. 43 (3). 799-804. | ||