Wiki des méthodes de biologie moléculaire et cellulaire
Glossaire collaboratif des méthodes expérimentales de biologie moléculaire et cellulaire. Ce glossaire est réalisé par les étudiants du module "Méthodes expérimentales de biologie moléculaire et cellulaire" sous la supervision de l'équipe de Génétique Animale.
Spécial | A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z | Tout
L |
|---|
| TK | La méthode de Bradford | ||
|---|---|---|---|
Quantification des protéines totales : La méthode de Bradford
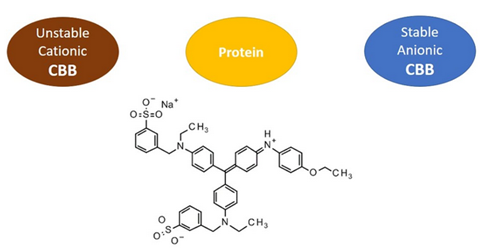
I) Objectifs La mesure de la concentration en protéines dans des échantillons est une partie essentielle de l’enzymologie ou de la biologie moléculaire. Ainsi, des méthodes de dosages rapides, sensibles et fiables sont nécessaire. Le dosage de protéine par la méthode de Bradford permet de mesurer la concentration totale de protéines dans un échantillon en utilisant le colorant bleu de Coomassie G-250. II) Principe Le dosage repose sur la liaison du colorant bleu de Coomassie G-250 aux protéines présentes dans l’échantillon, ainsi que sur son changement de couleur. Le colorant bleu de Coomassie change de couleur en fonction des paramètres du milieu : bleu sous sa forme anionique, vert à l’état neutre et rouge sous sa forme cationique (Figure 1).
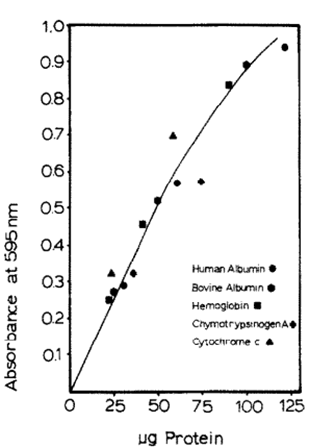
Figure 1 : Formule du bleu de Coomassie G-250 ainsi que la représentation de sa couleur en fonction de sa charge globale En solution acide, les protéines sont donc protonées, le colorant passe du rouge au bleu en se liant aux protéines à quantifier. Il forme des interactions hydrophobes et ioniques (Georgiou et al., 2008) avec les protéines. Cette liaison provoque un déplacement du maximum d’absorption de 465 à 595 nm, et c'est l'augmentation de l'absorbance à cette longueur d’onde qui est mesurée (Bradford, 1976). III) Mode opératoire Préparation du réactif protéique : Le Bleu de Coomassie G-250 est dissous dans de l’éthanol à 95 %, puis mélangé avec de l’acide phosphorique à 85 % pour former le réactif. Ce dernier est ensuite dilué jusqu’à un volume final de 1 litre, fournissant un réactif à concentration finale de 0,01 % de Coomassie G-250, 4,7 % d’éthanol, et 8,5 % d’acide phosphorique. Méthode standard : (pour des échantillons contenant de 10 à 100 µg de protéine) Des échantillons de protéines sont préparés dans un volume maximal de 0,1 ml, ajusté avec le tampon approprié. Un volume de 5 ml de réactif protéique est ajouté, et le mélange est homogénéisé par inversion ou vortexage. L’absorbance à 595 nm est mesurée après 2 minutes et avant 1 heure, dans des cuvettes de spectrophotomètre, contre un blanc de réactif préparé avec le même tampon et le réactif protéique. La relation entre la quantité de protéine et l’absorbance obtenue permet de construire une courbe d’étalonnage utilisée pour quantifier les protéines dans des échantillons inconnus. Méthode micro-protéine : (pour des échantillons contenant de 1 à 10 µg de protéine) Pour ces échantillons, le volume est ajusté à 0,1 ml avec le tampon approprié et un volume de 1 ml de réactif protéique est ajouté et mélangé. L'absorbance à 595 nm est mesurée, et une courbe d’étalonnage est utilisée pour quantifier la protéine. IV) Présentation des résultats Dans un premier temps, une gamme étalon doit être effectuée. Cette gamme est préparée avec des échantillons de différentes concentrations connues en protéines connues. L’absorbance est mesurée à 595 nm avec un spectrophotomètre (Figure 2).
Figure 2 : Résultat de la coloration d’une gamme d’étalonnage par la méthode de Bradford Ces valeurs sont intégrées dans un graphique décrivant l’absorbance à 595 nm en fonction de la masse de protéine de chaque échantillon de la gamme. Cette courbe permet de déterminer l’équation de la fonction linéaire reliant l’absorbance à 595 nm et la concertation en protéine. Cette équation est sous la forme : y = ax + b, A (595nm) = a x concentration en protéine + b (Figure 3).
Figure 3 : Résultat de mesure d’absorbance de différents échantillons de protéine avec la méthode Bradford. (Bradford, 1976) Ensuite, une fois cette équation obtenue, l’équation est appliquée aux échantillons dont la concentration en protéine est inconnue. Pour ce faire, il faut traiter ces échantillons avec le même réactif que la gamme et ensuite mesurer l’absorbance dans les mêmes conditions que la gamme d’étalonnage. Ainsi, une absorbance à 595 nm est obtenue pour ces échantillons.
V) Interprétation des résultats La concentration en protéines des échantillons inconnues est donc obtenue grâce à l’équation obtenue avec la gamme d’étalonnage en utilisant l’absorption mesurée de ces échantillons. Il faut cependant que l’absorbance mesurée pour ces échantillons soient compris dans le domaine de linéarité. La courbe d’étalonnage est linéaire sur une grande partie de la gamme. On peut donc mesurer des échantillons jusqu’à environ 100 µg de protéine. VI) Intérêts et limites Ce dosage, à la fois rapide et reproductible, permet une liaison quasi-instantanée du colorant aux protéines. Il se fixe au bout d’environ 2 minutes, avec une stabilité de la coloration pendant une heure. Il présente peu d’interférences avec les cations (Na⁺, K⁺) ou les glucides comme le saccharose. Bien que certains tampons alcalins puissent induire une légère coloration, l'utilisation de témoins appropriés permet d'assurer la précision de la mesure. Seules des concentrations élevées de détergents (SDS, Triton X-100) perturbent significativement l’analyse, tandis que les faibles quantités peuvent être neutralisées par des contrôles adéquats. VII) Références bibliographiques Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry 72: 248–254 Georgiou CD, Grintzalis K, Zervoudakis G, Papapostolou I (2008) Mechanism of Coomassie brilliant blue G-250 binding to proteins: a hydrophobic assay for nanogram quantities of proteins. Anal Bioanal Chem 391: 391–403
| |||

| AM | La minipréparation alcaline | ||
|---|---|---|---|
I) Objectifs Les plasmides bactériens sont fréquemment utilisés lors du clonage de gènes. La technique de la minipréparation permet d'extraire les plasmides des cellules bactériennes, afin de les identifier, de les caractériser et de les manipuler. Les nouveaux plasmides ainsi construits peuvent être isolés et caractérisés sur des gels d'électrophorèse. Il existe différentes méthodes de minipréparation, telles que la technique par ébullition ou la technique en une étape. La méthode par lyse alcaline est présentée ici. (Northrup et al., 2010) II) Principe La minipréparation (ou miniprep) est une technique de biologie moléculaire rapide et efficace, permettant d'extraire de petites quantités d'ADN (de l'ordre d'1 microgramme d'ADN par millilitre) de cultures bactériennes. Cette technique se base sur la dénaturation de l’ADN en fonction du pH. Il existe une étroite plage de pH, comprise entre 12 et 12,5, pour laquelle l’ADN linéaire est dénaturé, mais pas l’ADN circulaire. Grâce au pH basique, le lysat (acide) formé lors de la lyse des bactéries est neutralisé, et le pH reste stable afin de dénaturer uniquement l’ADN linéaire. (Birnboim et Doly, 1979)
III) Mode opératoire La première étape consiste à cultiver les cellules contenant le plasmide d'intérêt, durant 16 h à 40 h. Les bactéries sont ensuite lysées en milieu alcalin, dans une solution de SDS (dodécylsulfate de sodium) et de NaOH (hydroxyde de sodium), ce qui perturbe l'appariement des bases de l'ADN chromosomique et entraîne leur dénaturation. Après neutralisation du pH de la solution par ajout d’acétate de sodium ou de potassium, l'ADN plasmidique, qui est de petite taille (de l'ordre du kilobase) peut rapidement se renaturer, alors que l'ADN chromosomique ne le peut pas (sa taille est de l'ordre du mégabase). L’ajout de SDS dans le milieu entraîne également la précipitation du complexe SDS-protéines. Une étape de centrifugation sépare l’ADN chromosomique, l’ARN, les protéines et les débris cellulaires qui précipitent, de l'ADN plasmidique qui reste dans le surnageant. L’ADN est ensuite précipité par l'éthanol, conduisant à une quantité et à un degré de pureté d'ADN plasmidique suffisant pour la plupart des applications en laboratoire. Une des techniques fréquemment utilisées en laboratoire est la digestion de l'ADN plasmidique par une enzyme de restriction. L'ADN plasmidique isolé est analysé pour confirmer ou établir son identité. Les molécules d'ADN digérées sont séparées par électrophorèse sur gel d'agarose en fonction de leur taille. Pour améliorer cette étape, des enzymes RNases sont ajoutées afin de détruire l’ARN contaminant. (Birnboim et Doly, 1979; Northrup et al, 2010)
IV) Présentation des résultats L’ADN plasmidique récupéré grâce à la miniprep peut être analysé par électrophorèse après digestion par des enzymes de restriction. La figure 1 est une photo d'électrophorèse prise après la digestion par des enzymes de digestion d’un plasmide récupéré dans une bactérie par la méthode de la mini-prep alcaline (dans le cadre du clonage d’un gène). Le plasmide utilisé est le plasmide pgl3 d’une taille de 4 818 bp, et l’insert introduit est d’une taille de 471 bp. (Northrup et al., 2010)
Figure 1 : Photo d’électrophorèse du plasmide pgl3 contenu dans des bactéries après minipréparation alcaline et digestion enzymatique (UQAM, 2016)
V) Interprétation des résultats Sur l'électrophorèse, on peut voir 2 bandes, l’une à 5 000 bp qui correspond au plasmide, et l’autre à 500 bp qui correspond à l’insert. On peut conclure que la minipréparation à fonctionné et que le plasmide a bien été extrait de la bactérie.
VI) Intérêts et limites Les principaux intérêts de la minipréparation par lyse alcaline sont sa simplicité, son efficacité et son faible coût. Toutefois, cette technique connaît des limites. Les impuretés les plus retrouvées dans la solution d'ADN plasmidique purifié par miniprep sont les fragments d'ADN chromosomique. Du fait de sa taille supérieure à celle de l'ADN plasmidique, l'ADN chromosomique est fragmenté durant l'étape de lyse alcaline, ou par les forces de cisaillement qui apparaissent lors du pipetage. Ces fragments, de l'ordre du kilobase, précipitent avec l'ADN plasmidique, et peuvent se renaturer suite à la neutralisation de la solution. Bien que la minipréparation soit utilisée pour son efficacité et sa simplicité, l'ADN plasmidique n'est généralement pas assez pur pour être utilisé dans des techniques sensibles, telles que le séquençage d'ADN. Également, cette méthode implique que l’ADN plasmidique soit, comme l’ADN chromosomique, exposé à un pH alcalin. Or, une partie de l’ADN plasmidique subit une dénaturation irréversible à un pH supérieur à 13. (Felicello et Chinali, 1993; Chowdhury, 1991)
VII) Références bibliographiques
HC. Birnboim, J. Doly (1979). A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Research, Volume 7, pages 1513-1523.
I. Felicello, G. Chinali (1993). A modified alkaline lysis method for the preparation of highly purified plasmid DNA from Escherichia Coli. Analytical biochemistry. Volume 2012 (tome 2), pages 394-401.
J. Lemoine, UQAM (2016). Photo électrophorèse du plasmide pgl3 contenu dans des bactéries après minipréparation alcaline et digestion enzymatique (2016). Unpublished
K. Chowdhury (1991). One step “miniprep” method for the isolation of plasmid DNA. Nucleic Acids Research. Volume 19, n°10, page 2792.
VA. Northrup, CJ. Backhouse, DM. Glerum (2010). Development of a microfluidic chip-based plasmid miniprep. Analytical Biochemistry. Volume 402, n°2, pages 185-190.
| |||

| CP | Le Compteur Coulter | |
|---|---|---|
Compteur CoulterI)
Objectifs La méthode Coulter est une méthode de dénombrement qui permet de compter des particules microscopiques en suspension dans un électrolyte, en fonction de leur taille. (GRAPPIN, R. et JEUNET, R., 1971). D'après BECKMAN COULTER, 2023, la méthode a été inventée à la fin des années 40 par un ingénieur de la Navy aux Etats-Unis pour compter les cellules sanguines rapidement et précisément, marquant la naissance de l’hémogramme. II) Principe Depuis les années 50, la méthode Coulter a été utilisée pour la caractérisation de milliers de matériel biologiques et industriels : levures, bactéries, cellules sanguines, drogues, pigments, aliments, abrasifs, explosifs, minéraux, métaux, argile. Le principe de Coulter repose sur la mise en suspension de cellules ou particules comprises entre 0.2 µm et 1600µm de diamètre dans un électrolyte (liquide conducteur). La solution analysée est mise dans une cuve dans laquelle sont positionnées deux électrodes entretenant le courant électrique. (FINOT. E., 2010) L'une d'entre elles est située dans la sonde en verre contenant un petit orifice cylindrique calibré. Les particules passant par l'ouverture déplacent un volume de solution électrolytique égal à leur propre volume. Ce déplacement entraîne une modification de la résistance électrique à travers l'ouverture, ce qui provoque une impulsion électrique. Par conséquent, le nombre d’impulsions électriques provoquées est proportionnel au nombre de cellules tandis que l’intensité de l’impulsion est proportionnelle au volume des particules. (BECKMAN COULTER, 2023) Diverses études utilisent cette méthode dans le monde entier telles que : l’étude expérimentale d’algues unicellulaires, la détermination du nombre de cellules totales du lait de troupeau, l’effet antibactérien sur une croissance de micro-organismes. (ROBERT, R., 1985 ; KEBBAL, S., GHARBI, I. et al, 2008) III) Mode opératoireTout d'abord, un volume de la solution contenant les cellules en suspension est placé dans la cuve. Un courant
électrique y est ensuite appliqué. Les cellules présentes dans le volume de la
solution vont passer une à une par l’orifice en raison du courant électrique
appliqué, allant de l'anode vers la cathode. Leur passage au travers de
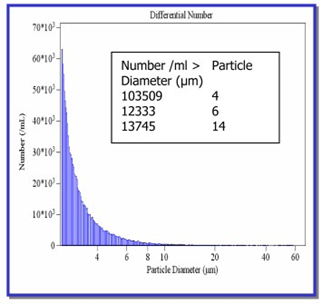
l’orifice provoque un changement de courant électrique q 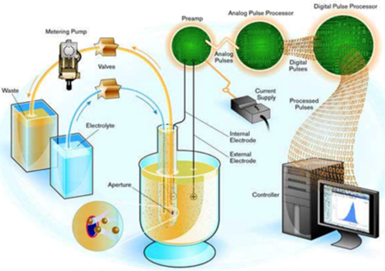 Figure 1 : Schéma représentatif de la méthode Coulter. (BECKMAN COULTER, 2023) IV) Présentation des résultatsGrâce à l’appareil, un graphique du nombre de particules en fonction de leur diamètre est obtenu. Il permet de connaître la répartition des cellules dans la solution, leur nombre ainsi que leurs dimensions. Chaque impulsion correspond à une cellule. Sur la figure 2, le graphique rouge et le graphique bleu représentent deux échantillons de mitochondries de concentrations différentes. (BECKMAN COULTER, 2023) Quant à la figure 3, un seul échantillon est analysé. (AZO NETWORK, 2014)
Figure 2 : dénombrement cellulaire (/ml) en fonction du diamètre des cellules (µm) par le Multisizer4e. (BECKMAN COULTER, 2023)
Figure 3 : dénombrement cellulaire (/ml) en fonction du diamètre des cellules (µm) par le Multisizer3. (AZO NETWORK, 2014) V) Interprétation des résultatsLa figure 2 présente 2 graphiques obtenus de couleurs différentes correspondent à 2 échantillons différents. Le graphique rouge présente une concentration cellulaire plus importante que l’échantillon bleu. Également, les cellules de l’échantillon rouge sont de plus grandes tailles. Puis, pour la figure 3, l’encadré présente le nombre total de particules, ainsi que la distribution du nombre et des tailles de celles-ci en fonction d’une classification. Sur les 2 figures (figures 2 et 3), plus la taille des cellules augmente, plus le nombre de cellules par mL diminue. (BECKMAN COULTER, 2023 ; AZO NETWORK, 2014) VI) Intérêts et limitesCette méthode comporte plusieurs avantages : elle permet de dénombrer un très grand nombre de particules différentes (bactéries, levures, mitochondries, cellules sanguines, …), peu importe leurs caractéristiques physiques (forme, couleur), et près de 10 000 particules par seconde peuvent être dénombrées de taille comprise entre 0,4 et 1 600 µm. Ainsi, cette méthode donne des résultats très précis. Également, l’obtention de la distribution des tailles peut être réalisée en un temps relativement court. L’automatisation de la méthode permet une grande rapidité et une limitation des erreurs liée au comptage manuel. (KHALIFA M. A., 2021) Cependant, l’instrumentation nécessaire représente un budget important et cette méthode ne peut pas s’appliquer à toutes les particules. En effet, la méthode Coulter ne peut être utilisée pour les particules de trop petite taille et qui ne peuvent pas être mises en suspension dans une solution électrolytique. Le comptage est dépendant de l’orifice sélectionné ; un mauvais choix induira un résultat biaisé, car les particules ne pourront passer ou passeront trop vite et seront mal comptées par l’appareil. (BECKMAN COULTER, 2023) VII) Références bibliographiquesAZO NETWORK, (2014). Using the MultisizerTM 3 Coulter Counter to Determine the Size and Concentration of Particles in Oil. AZoM.com. Disponible à l’adresse : https://www.azom.com/article.aspx?ArticleID=11501 BECKMAN COULTER, (2023). Coulter Principle, Counting and Sizing Particles. Disponible à l’adresse : https://www.beckman.fr/resources/technologies/flow-cytometry/history/coulter-principle BHATTACHARJEE. S (2019). Coulter Counter. Disponible à l’adresse : KHALIFA M. A., (2021). Coulter Counter Method | Conductivity Method of particle analysis | Micromeritics | Animation. Disponible à l’adresse : FINOT. E., (2010). Electrocinétique 2.3 : Compteur de Coulter. Disponible à l’adresse : GRAHAM, M., (2013). The Coulter principle: Imaginary origins. Cytometry. Vol. 83, (n° 12), pp. 1057‑1061. DOI 10.1002/cyto.a.22398. GRAPPIN, R. et JEUNET, R., (1971). Essais de l’appareil " Compteur Coulter " utilisé pour la détermination du nombre de cellules totales des laits de troupeaux. Le Lait. Vol. 51, (n° 505‑506), pp. 273‑293. DOI 10.1051/lait:1971505-50615. KEBBAL, S., GHARBI, I., GUEMRA, S., HANZEN, C. et GUETARNI, D., (2008). Validation d’une méthode de dénombrement de la concentration en cellules somatiques du lait de vache au moyen du Coulter Counter® modèle Z2. Annales de Médecine Vétérinaire. Vol. 152, (n° 4) Disponible à l’adresse : https://orbi.uliege.be/handle/2268/59670 ROBERT, R., (1985). Intérêt du compteur de particules ZB-ZBI et de l’analyseur C1000 pour la numération des algues unicellulaires de culture. Revue des Travaux de l’Institut des Pêches Maritimes. Vol. 49, (n° 3‑4), pp. 155‑163. | ||

| PB | Le Pyroséquençage | |
|---|---|---|
PYROSEQUENCAGE
I) Objectifs Le pyroséquençage est une technique développée à partir de 1985 (1), permettant le séquençage de l’ADN (2). Cette méthode est principalement utilisée pour analyser les SNP (Single-nucleotide polymorphisms) et ainsi permettre l'étude de la variabilité génétique et la détection d’haplotypes. Le pyroséquençage permet l'étude de la fréquence allélique dans de grandes populations, étant donné que les signaux obtenus par cette technique sont très quantitatifs. Une autre application de cette technique est aussi l’identification de bactéries, champignons, et de virus en vue de découvrir de nouvelles espèces, de nouveaux groupes taxonomiques, via le séquençage de l’ARNr 16S (1).
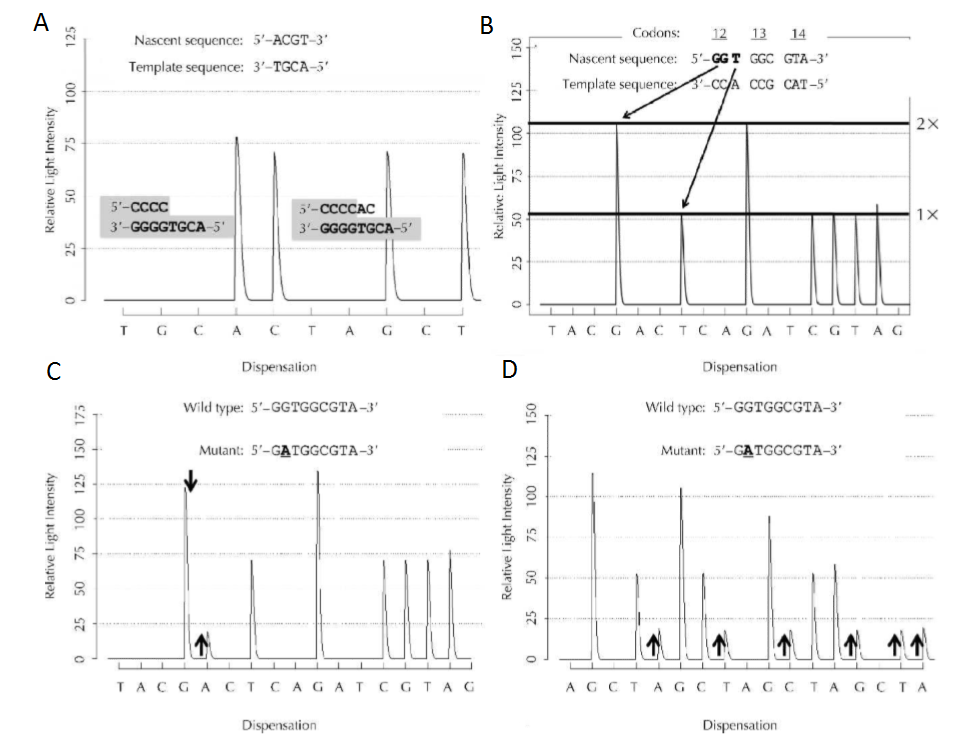
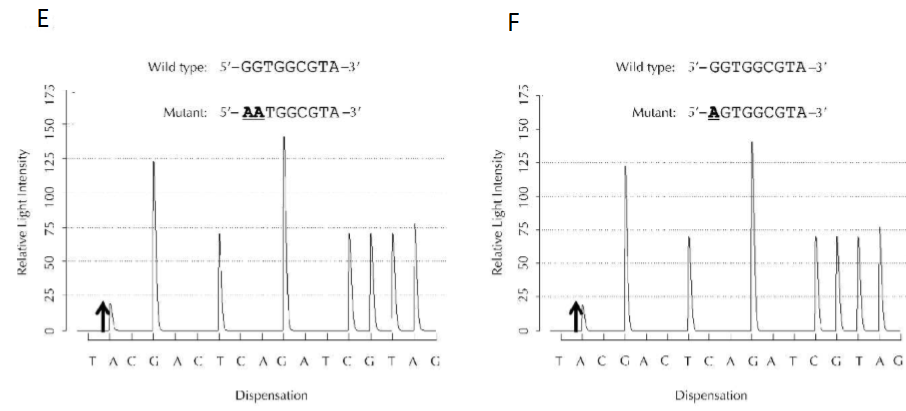
II) Principe Cette technique est basée sur le principe de séquençage par synthèse, qui s’oppose au séquençage par “terminaison” qui est utilisé dans la méthode de Sanger. Le pyroséquençage repose sur une cascade de réactions (Figure 1). :
Figure 1: Principe du pyroséquençage (Ronaghi, 2001) III) Mode opératoire Avant d’effectuer le pyroséquençage, une PCR est réalisée de manière à obtenir un produit PCR biotinylé. Ce produit est ensuite des billes de Sepharose enrobées de streptavidine. Les billes sont lavées et dénaturé, ce qui permet d’obtenir de l’ADN simple brin adapté au pyroséquençage. L’ADN matrice est mis dans la plaque de réaction de pyroséquençage contenant l’amorce. Une fois que l’amorce s’est hybridée sur le brin d’ADN, la plaque est placée dans la machine de pyroséquençage PyroMark. Au cours du pyroséquençage, les réactifs (enzymes, nucléotides, substrat) sont ajoutés séquentiellement et à des volumes fixés. L’ensemble du processus est régi par un logiciel qui permet à la machine d’assurer les différentes étapes. La machine Pyromark permet de séquencer simultanément jusqu’à 24 échantillons en moins de 15 minutes ().
IV) Présentation des résultats Le capteur Charged Coupled Device (CCD) capte le signal lumineux émis par les luciférases, et le traduit par un pic sur le pyrogramme. La figure 2 montre l’intensité lumineuse relative détectée par le capteur à l’ajout du nucléotide indiqué en abscisse. Il est ainsi possible de connaître la composition de la séquence observée. Voici quelques exemples de pyrogrammes que nous expliquerons dans la partie V).
Figure 2: Exemples de pyrogrammes (Harrington et al., 2013)
V) Interprétation des résultats Pour chaque salve de nucléotides correctement appariés, un pic est obtenu. Lorsque le nucléotide proposé n'est pas le bon, aucun signal n'est détecté. La hauteur de chaque pic est fonction de l'intensité du signal, et proportionnelle au nombre de nucléotides identiques détectés à la suite. Par exemple, dans le graphique A de la Figure 2, le brin codant comporte une Thymine et une Guanine à la suite. Ainsi, la proposition d’une Adénine, puis d’une Cytosineentraine l’émission de deux pics successifs de tailles identiques. Ensuite, étant donné que les bons nucléotides ne sont pas proposés (Thymine, Adénine), il n’y a pas d’émission lumineuse. Sur le graphique B de cette même figure, nous observons que la présence de deux Cytosines d’affilée entraîne la création d’un pic Guanine à l’intensité relative deux fois supérieure aux pics “simples”. A partir du schéma du signal, la séquence étudiée peut être déduite. La distribution de nucléotides sur le pyrogramme peut prendre deux formes : la distribution cyclique, ou la distribution optimisée. La distribution cyclique (Fig.2, graphique D) consiste à toujours proposer les nucléotides dans le même ordre (par exemple : AGCTAGCTAGCT...). La distribution optimisée (Fig.2, graphique C) n'a pas d'ordre particulier, mais est adaptée à la séquence étudiée. Or, la distribution cyclique ne permet pas de détecter les mutations complexes, ce qui risque de donner une séquence fausse. Les mutations complexes sont caractérisées par des mutations impliquant plus de deux bases substituées, et pas forcément consécutives. En revanche, elle est la seule option adaptée pour séquencer une cible inconnue, comme par exemple un génome entier. Ces deux techniques mènent donc à des pyrogrammes différents, bien que les séquences soient les mêmes (3).
Cette technique est utilisée dans le cadre de la détection de mutations (transformant ainsi des gènes sains en oncogènes, qui sont des gènes impliqués dans le développement de tumeurs), l’analyse de méthylation, ou encore l’analyse de résultats complexes issus de la méthode de Sanger. Dans le même temps, les pyrogrammes du Wild Type et du mutant sont réalisés, afin de repérer en temps réel les mutations entre les deux individus, sur la région d’intérêt. Les intérêts de cette technique sont nombreux. Celle-ci ne nécessite aucun clonage, ce qui entraîne des gains de temps et aussi d'argent. Le pyrogramme peut être étudié en temps réel, ce qui permet la lecture directe de la séquence obtenue, et détecte plus facilement les allèles mutants que la méthode de Sanger. En revanche, certains aspects pourraient encore être optimisés. Par exemple, si trop de nucléotides identiques se suivent, il devient difficile de les quantifier précisément. En effet, la taille du pic devient disproportionnée en comparaison aux autres pics environnants. De plus, le pyroséquençage ne lit que des séquences relativement courtes en comparaison avec la méthode de Sanger.
VII) Références bibliographiques 1 -AHMADIAN, Afshin, EHN, Maria et HOBER, Sophia, (2006). Pyrosequencing: History, biochemistry and future. In : Clinica Chimica Acta. janvier 2006. Vol. 363, n° 1‑2, p. 83‑94. DOI 10.1016/j.cccn.2005.04.038. 2 -HARRINGTON, Colleen T., LIN, Elaine I., OLSON, Matthew T. et ESHLEMAN, James R., (2013). Fundamentals of pyrosequencing. In : Archives of Pathology and Laboratory Medicine. 2013. Vol. 137, n° 9, p. 1296–1303. 3 - F. SANGER, S. NICKLEN et A.R. COULSON, « DNA sequencing with chain-terminating inhibitors », Proc. Natl. Acad. Sci. USA, vol. 74, 1977, p. 5463-5467
4-RONAGHI, Mostafa, (2001). Pyrosequencing sheds light on DNA sequencing. In : Genome research. 2001. Vol. 11, n° 1, p. 3–11. 5-https://www.qiagen.com/us/shop/automated-solutions/sequencers/pyromark-q24/#productdetails
| ||

| RB | Les Méthodes ELISA | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Les Méthodes ELISA
I) Objectifs ELISA (Enzyme Linked ImmunoSorbent Assay) est une méthode immuno-enzymatique permettant la détection d’un antigène dans un échantillon. Elle a été mise au point en 1971 par Engvall et Perlmann. Elle a notamment beaucoup servi durant la pandémie de la COVID-19.
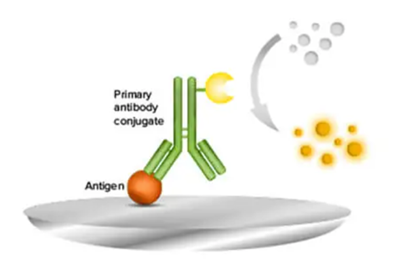
II) Principe La méthode repose sur un dosage immunologique dans un échantillon permettant de savoir s’il contient ou non un antigène. La méthode ELISA se décline en quatre versions :
III) Mode opératoire Test ELISA Direct :
Test ELISA Indirect :
Test ELISA Sandwich :
Test ELISA Compétitif :
Figure 1 : Schémas des mécanismes de capture des différentes méthodes ELISA (Molecular Devices - Dosage d’immunoabsorption enzymatique)
IV) Présentation des résultats Les résultats sont directement observables sur la plaque ELISA lorsqu’elle est qualitative. En revanche, si l’objectif est d’avoir une ELISA quantitative, il est nécessaire de présenter les résultats sur une courbe d’étalonnage, ou via une densité optique (DO).
Figure 2 : Photo d’une plaque 96 puits de résultats ELISA sandwich
V) Interprétation des résultats Le résultat est interprété différemment dans le cas où l’ELISA est quantitative ou qualitative. Lorsqu’elle est quantitative, le calcul de la concentration de l’antigène dans l’échantillon est calculé à partir d’une courbe d’étalonnage avec comme paramètre la densité optique ou les unités de fluorescence. En revanche, si l’ELISA est qualitative, le résultat indiquera simplement la présence ou l’absence de l’antigène dans l’échantillon d’étude.
VI) Intérêts et limites
VII) Références bibliographiques Olsen C (2020) Histoire du test ELISA, de sa création à la recherche sur la COVID-19. https://fr.moleculardevices.com/lab-notes/microplate-readers/the-history-of-elisa Diagomics Les différentes techniques ELISA. https://www.diagomics.com/methode-elisa Spirckel P (2024) Méthode ELISA (Enzyme-Linked Immuno Assay). https://wiki.fablab.sorbonne-universite.fr/BookStack/books/techniques-de-base/page/methode-elisa-enzyme-linked-immuno-assay Dosage d’immunoabsorption enzymatique (ELISA). Molecular Devices, https://fr.moleculardevices.com/applications/enzyme-linked-immunosorbent-assay-elisa Engvall E, Perlmann P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry. 1971 Sep;8(9):871-4. doi: 10.1016/0019-2791(71)90454-x. PMID: 5135623.
| |||||||||||||||||||||




| EL | Live-seq | ||
|---|---|---|---|
Live-seq : Méthode de séquençage du transcriptome de cellules uniques
I) Objectifs La méthode Live-seq est une méthode de séquençage du génome très peu invasive. Cette méthode a pour objectif de préserver les cellules pour permettre de suivre l’activité des gènes dans le temps (Papageorgiou, 2022). Elle repose sur la microscopie à force fluidique. L’objectif du Live-seq est le séquençage du transcriptome de cellules uniques, plus précis que le séquençage de tissus composés de plusieurs types cellulaires (Schutz, 2017). II) Principe Cette nouvelle technologie de séquençage consiste à extraire de l’ARN messager (ARNm) d’une cellule sans le détériorer, en utilisant un microscope à force fluidique. Le profilage du transcriptome peut être alors obtenu par séquençage ARN tout en maintenant la cellule en vie (méthode de RNA-seq) (Papageorgiou, 2022). III) Mode opératoire 1. La microscopie à force fluidique En microscopie à force fluidique, la cellule est isolée et encapsulée dans une gouttelette d’huile contenant le GEM (Gel bead in EMulsion) grâce à un système combinant la microscopie à force atomique et des micro canaux à force fluidique extrêmement fins. Cette gouttelette d’huile GEM est une bille composée : - D’une séquence Poly T, qui grâce à sa complémentarité à la queue poly A, va pouvoir se fixer aux ARNm. - De plusieurs UMI, étiquettes moléculaires permettant de détecter des erreurs d’amplifications, - D’un bar-code, qui est unique à la bille en question. Ce bar-code, également unique à la cellule, permet une identification des ARNm (Schutz, 2017). La technique d’extraction appelée FluidM consiste à prélever de très petite quantité cytoplasmique et ainsi prélever l’ARNm cytoplasmique sans détruire la cellule (Chen et al., 2022). Le but est de minimiser la perte ou la dégradation de l’ARNm prélevé (Figure 1). Figure 1 (Chen et al., 2022) : Procédure d'échantillonnage Live utilisant FluidM (ici, appliquée sur des cellules préadipocytes brunes IBA). Les flèches blanches indiquent l'application d'une sous-pression ou d'une surpression. Les flèches noires indiquent la quantité de tampon et d'extrait dans la sonde. Barre d'échelle, 20 μm. (1) Pré Chargement du tampon, (2) Ciblage de la cellule, (3) Extraction et (4) Libération de l’extrait 2. Réaction de RNA-seq : la méthode Smart-seq La méthode de séquençage de base utilisée est le Smart-seq (Figure 2). Une fois l’ARNm récupéré et polyadénylé, il est possible de le transcrire en ADNc via l’intervention d’une reverse transcriptase en présence de TSO (Template Switch oligo). Ce TSO est ensuite clivé de manière enzymatique (méthode du template switching). Par la suite, une PCR est utilisée afin d’amplifier cet ADNc. L’ADN amplifié est ensuite incubé avec des transposases afin de fragmenter les transcriptions complètes et d’y ajouter des adaptateurs (Trombetta et al., 2018).
Le protocole a ensuite été adapté pour que 1 pg d’ARN total soit détectable. Ceci représente environ 10 % de l’ARN cellulaire total (Chen et al., 2022) Enfin, après purification, chaque bibliothèque unicellulaire obtenue est codée par PCR et peut être séquencée par la méthode illumina (Schutz, 2017). 3. Création d’une bibliothèque Après séquençage par illumina de l’échantillon, une librairie de gènes est créée. En analysant la composition en gènes des différentes cellules échantillonnées, une matrice d’expression est obtenue (Figure 3) et peut être représentée dans un graphique (Schutz, 2017).
IV) Présentation des résultats Par une analyse statistique d’analyse en composante principale, la méthode Live-seq permet d’obtenir un graphique où chaque point correspond à une cellule. Plus les points sont éloignés et plus ils possèdent des caractéristiques génétiques différentes (Schutz, 2017).
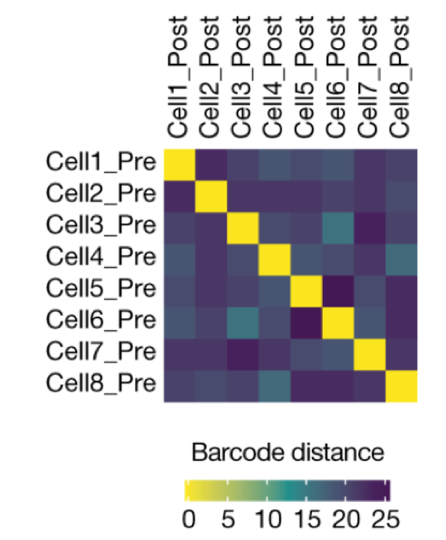
Figure 4 (Schutz, 2017) : Profil d’expression obtenu à partir de cellules du sang (après clusterisation des familles) Différentes représentations peuvent être réalisées en fonction de l’objectif de l’expérience. Dans l’exemple ci-dessous, les résultats sont présentés dans une carte montrant la distance des codes-barres de suivi dans les cellules échantillonnées afin de pouvoir les comparer (École Polytechnique Fédérale de Lausanne, 2022).
V) Interprétation des résultats Les résultats peuvent être interprétés de différentes manières en fonction de l’objectif de l’expérience. Par exemple, le séquençage du transcriptome de la cellule peut être réalisé à plusieurs instants, permettant ainsi de suivre son évolution au cours du temps. Une comparaison des différentes cellules échantillonnées est également possible dans divers domaines d’application (notamment la médecine) (Chen et al., 2022). VI) Intérêts et limites Cette méthode permet en premier lieu de stratifier différents types et états cellulaires de divers échantillons. La cellule ayant été préservée lors de l’extraction de l’ARNm à l’instant t, il est donc possible de prélever à nouveau du matériel génétique au sein de la même cellule à l'instant t+1. L’évolution temporelle du transcriptome de la cellule est alors obtenue permettant ainsi de mieux comprendre les relations entre comportement moléculaire et phénotypique à un instant précis : il s’agit d’une analyse transcriptomique “temporelle” (École Polytechnique Fédérale de Lausanne, 2022). Toutefois, la procédure de Live-seq n’est encore utilisée que pour des cultures cellulaires. Des travaux sont en cours pour pouvoir étendre sa capacité à d’autres champs d’application. Enfin, augmenter la capacité de détection de l’ARNm ou du pouvoir de résolution cellulaire permettraient d'accroître l'efficacité de la méthode (Chen et al., 2022). VII) Références bibliographiques Chen W, Guillaume-Gentil O, Rainer PY, Gäbelein CG, Saelens W, Gardeux V, Klaeger A, Dainese R, Zachara M, Zambelli T, et al (2022) Live-seq enables temporal transcriptomic recording of single cells. Nature 608: 733–740 École Polytechnique Fédérale de Lausanne (2022) Live-seq- sequencing a cell without killing it. RNA-seq Papageorgiou N (2022) Live-seq: séquencer sans tuer la cellule. Sciena, https://www.sciena.ch/fr/research/liveseq-sequencing-a-cell-without-killing-it.html Schutz S (2017) Séquençage des ARNm sur cellules uniques. Sacha Schutz : bioinformatique génétique médecine, https://dridk.me/sc-rna-seq.html Trombetta JJ, Gennert D, Lu D, Satija R, Shalek AK, Regev A (2018) Smart-seq2 single-cell RNA-Seq modified method. Protocols.io
| |||