Wiki des méthodes de biologie moléculaire et cellulaire
Glossaire collaboratif des méthodes expérimentales de biologie moléculaire et cellulaire. Ce glossaire est réalisé par les étudiants du module "Méthodes expérimentales de biologie moléculaire et cellulaire" sous la supervision de l'équipe de Génétique Animale.
Spécial | A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z | Tout
R |
|---|
| CH | Recombineering | |
|---|---|---|
Recombineering
I) Objectifs Le Recombineering (homologous RECOMBInation-mediated genetic engiNEERING) est une technique de génie génétique permettant de créer des modifications génétiques précises in vivo. Elle est basée sur des systèmes de recombinaison homologue. Cette méthode peut être utilisée pour la construction de knock-out, de délétions, d'insertions, d’inversions, de mutations ponctuelles, de remplacements de gènes. Le clonage de gènes, la construction de plasmides in vivo et le marquage de gènes/protéines sont également possibles (Sawitzke et al., 2023). II) Principe Le recombineering utilise des substrats d’ADN linéaires qui peuvent être double-brin (dsDNA), généralement sous la forme de produits PCR, ou simple-brin (ssDNA) (oligonucléotides synthétiques). Ces substrats contiennent la modification que l’on souhaite apporter et sont flanqués de courtes séquences homologues à l’ADN cible (30-50 bases). Ces substrats sont introduits dans des cellules exprimant des enzymes de recombinaison, pouvant être codées par des bactéries, bactériophages ou levures. Ces enzymes incorporent l’ADN linéaire dans la séquence cible, donnant ainsi des molécules recombinantes. III) Mode opératoire Il existe différents modes opératoires du recombineering, dépendants du but recherché. Nous allons prendre ici comme exemple l’insertion d’un marqueur sélectionnable à l’aide du système de recombineering le plus utilisé, celui du bactériophage λ Red. Ce système est composé de 3 protéines : Gam, Exo et Beta. Tout d’abord, l’ADN linéaire contenant la modification à apporter et les séquences homologues à l’ADN cible est généralement introduit dans les cellules par électroporation. Une fois dans la cellule, la protéine Exo se fixe sur l’ADN linéaire. Exo est une exonucléase 5′→3′ spécifique de l'ADN double brin qui va couper l’ADN de 5’ vers 3’, laissant des surplombs 3’ de chaque côté de l’ADN. La protéine β va se fixer sur l’ADN simple brin (les surplombs laissés) et favoriser l’appariement de ce brin d’ADN avec l’ADN cible homologue (figure 1) (Sawitzke et al., 2013a). La protéine Gam inhibe l'exonucléase RecBCD d'E. coli, qui dégrade normalement l'ADNdb linéaire. Gam n'est pas absolument nécessaire à la recombinaison, mais elle multiplie par 20 la fréquence de la recombinaison de l'ADNdb. Les clones recombinants sont sélectionnés (ex: sélection de clones antibiorésistants si le marqueur sélectionnable était un gène d’antibiorésistance) et confirmés par PCR (Sawitzke et al., 2013a).
Figure 1 : Schéma de l’insertion d’un marqueur sélectionnable à l’aide du système de recombineering du bactériophage λ Red
Ceci n’est qu’un exemple des modes opératoires possibles pour le recombineering. Chaque mode opératoire comporte cependant les six étapes suivantes (figure 2) (Sharan et al., 2009): - Génération du substrat d'ADN linéaire : ADN double-brin ou simple-brin- Mise à disposition des gènes de recombinaison : les gènes de recombinaison peuvent être introduits de différentes manières dans les cellules bactériennes où la recombinaison sera effectuée. - Induction des gènes de recombinaison : dans le cas du système de recombinaison λ Red, celui-ci est induit en incubant la culture bactérienne à mi-log dans un bain-marie à 42°C en agitant à 200 tours par minute pendant 15 minutes. Immédiatement après l'impulsion thermique, les cellules doivent être placées dans une bouillie d'eau glacée pour un refroidissement rapide. - Préparation de cellules électrocompétentes et électroporation du substrat d'ADN - Croissance après électroporation - Identification et confirmation des clones recombinants
Figure 2 : Représentation schématique des différentes étapes du recombineering IV) Présentation des résultats La présentation des résultats varie selon l’objectif de la manipulation. Pour démontrer l’efficacité du recombineering, une PCR, une digestion par des enzymes de restriction, une analyse par Southern Blot ou un séquençage peuvent être réalisés (Sharan et al., 2009). V) Interprétation des résultats Ici aussi l’interprétation des résultats dépend de la méthode choisie. Les recombinants antibio-résistants sont d'abord sélectionnés par un test d’antibiorésistance approprié, puis analysés par PCR pour vérifier que l'insertion s'est faite au bon endroit et que les cellules ne sont pas diploïdes pour le locus. Dans certains cas, comme pour les plasmides à copies multiples, une analyse de restriction peut être utilisée pour confirmer les clones recombinants. Les Southern blots et le séquençage peuvent être utilisés pour analyser les recombinants BAC ou génomiques. En cas de mutation ponctuelle ou d'autre changement subtil, le séquençage de l'ADN doit être utilisé pour confirmer l'exactitude des recombinants (Sharan et al., 2009). VI) Intérêts et limites Intérêts : - Le recombineering permet d'apporter des modifications génétiques rapides, précises et peu coûteuses à n'importe quelle séquence d'ADN (Sawitzke et al., 2013b)- Le recombineering étant basé sur des homologies entre séquences, il permet de modifier n’importe quelle séquence avec précision sans avoir besoin de sites de restriction, contrairement aux autres méthodes de génie génétique (Sharan et al., 2009). - Cette méthode permet également de faciliter les altérations génétiques sur de grandes molécules d’ADN. En effet, dans de grandes molécules, il peut être difficile de trouver des sites de restrictions uniques, ce qui complique la réalisation de ces tâches via les techniques de génie génétique classiques. Hors, comme dit précédemment, le recombineering peut se faire indépendamment des sites de restriction (Sawitzke et al., 2023).
Limites : - Étant donné que de très courtes régions d'homologie suffisent pour la recombinaison, la manipulation de régions contenant des séquences répétitives peut s'avérer problématique (Sharan et al., 2009).- Les produits recombinants peuvent occasionnellement subir des mutations, puisque la plupart des méthodes de recombineering se basent sur des produits amplifiés par PCR comme ADN substrat. Cependant, cela peut se vérifier par séquençage (Sharan et al., 2009). - La séquence de la région cible doit être connue. Cependant, comme le génome entier de nombreux organismes a été séquencé, cela n'est pas une limitation pour la plupart des organismes couramment utilisés (Sharan et al., 2009).
VII) Références bibliographiques Sawitzke JA, Barenghi A, Thomason L, Costantino N, Court D (2023). Recombineering: A Modern Approach to Genetic Engineering. Reference Module in Life Sciences. doi: 10.1016/B978-0-12-822563-9.00100-1 Sawitzke JA, Thomason LC, Bubunenko M, Li X, Costantino N, Court DL (2013a). Chapter Seven - Recombineering: Using Drug Cassettes to Knock out Genes in vivo. In J Lorsch, ed, Methods in Enzymology. Academic Press, pp 79–102 Sawitzke JA, Thomason LC, Bubunenko M, Li X, Costantino N, Court DL (2013b).Chapter Ten - Recombineering: Highly Efficient in vivo Genetic Engineering using Single-strand Oligos. In J Lorsch, ed, Methods in Enzymology. Academic Press, pp 157–177 Sharan SK, Thomason LC, Kuznetsov SG, Court DL (2009). Recombineering: A Homologous Recombination-Based Method of Genetic Engineering. Nat Protoc 4: 206–223 Céline Heel, Manon Rineau, 2024 | ||


| LN | Restriction Fragment Length Polymorphism (RFLP) | |
|---|---|---|
Restriction Fragment Length Polymorphism (RFLP)
Objectifs La Restriction Fragment Length Polymorphism (RFLP) est une méthode d’analyse génétique utilisée pour la première fois en 1980 par Botstein et al. Elle permet de détecter le polymorphisme génétique inter- ou intra- espèce dans les régions codantes et non codantes (Rasmussen, 2012).
Figure 1 : Principe de la RFLP. Source : d’après (NCBI, 2017) 1- Extraction de l’ADN génomique 2- Amplification de l’ADN génomique par PCR 3- Vérification de l’amplification par électrophorèse suivie d’une révélation 4- Digestion enzymatique de l’ADN 5- Séparation des fragments d’ADN par électrophorèse Les fragments d’ADN de différentes tailles sont séparés par électrophorèse. 6- Révélation des fragments de la séquence analysée
NCBI (2017). Restriction Fragment Length Polymorphism (RFLP). Dans : NCBI. [Consulté le 27 mars 2018]. Disponible à l’adresse: https://www.ncbi.nlm.nih.gov/probe/docs/techrflp/. | ||


| PG | Ribo-Sequencing (Ribo-seq) | |
|---|---|---|
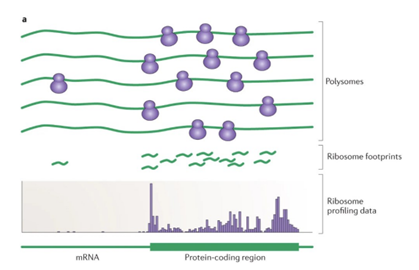
I) Objectifs Le Ribo-Seq est une technique permettant d’analyser la traduction des ARNm à une résolution nucléotidique en identifiant les fragments d’ARN protégés par les ribosomes (Ingolia et al., 2009), cette approche offre une vision détaillée de l’expression génique au niveau translationnel et permet d’étudier les cadres de lecture ouverts (Open reading frame, ORF) traduits, la régulation de la traduction et l’efficacité traductionnelle des transcrits. II) Principe Le Ribo-Seq repose sur la digestion enzymatique des ARNm non protégés par les ribosomes, suivie de l’extraction et du séquençage des fragments restants, appelés "footprints". L’alignement de ces séquences sur le génome permet d’obtenir un profil précis de l’activité traductionnelle des cellules. Cette approche a considérablement amélioré notre compréhension de la régulation de la traduction, notamment grâce aux travaux d’Ingolia (2016) et de McGlincy & Ingolia (2017).
III) Mode opératoire
Figure 1 : Illustration des étapes du Ribo-Sequencing.
La méthode suit plusieurs étapes (Figure 1): 1. Lyse cellulaire pour figer la traduction en cours. 2. Digestion des ARNm non protégés à l’aide d’une RNase spécifique. 3. Purification des fragments ribosomaux pour isoler les ARNm protégés par les ribosomes. 4. Construction d’une bibliothèque d’ADNc en vue du séquençage. 5. Séquençage haut débit et analyse bioinformatique pour cartographier les sites de traduction actifs.
Figure 2 : Illustration du principe du Ribo-Seq et la dérivation des DRs correspondantes (Ingolia, 2014). IV) Présentation des résultats Les résultats sont souvent représentés sous forme de profils de densité ribosomale alignés sur un génome de référence. L’identification des ORF traduits repose sur la distribution des footprints ribosomiques et permet d’observer l’efficacité traductionnelle des transcrits. V) Interprétation des résultats L’analyse des profils ribosomaux permet de quantifier l’impact de divers mécanismes régulateurs sur la traduction. Des études ont révélé l’existence de cadres de lecture alternatifs, de pauses ribosomales et de sites d’initiation non canoniques, notamment dans des situations de réponse au stress (Ingolia, 2016). VI) Intérêts et limites Le Ribo-Seq apporte une résolution inégalée pour l’étude de la traduction, surpassant les méthodes traditionnelles basées sur le RNA-Seq. Il permet d’étudier des mécanismes fondamentaux de régulation translationnelle et a contribué à la découverte de nouveaux peptides codés par des ORF auparavant non identifiés. Toutefois, la méthode présente certaines limites, notamment la nécessité de contrôles rigoureux pour éviter les biais expérimentaux et la complexité des analyses bioinformatiques requises (McGlincy & Ingolia, 2017). VII) Références bibliographiques Ingolia, N.T., Ghaemmaghami, S., Newman, J.R., & Weissman, J.S. (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science, 324(5924), 218-223.
Ingolia, N.T. (2016). Ribosome profiling : New views of translation, from single codons to genome scale. Nature Reviews Genetics, 17(3), 155-167.
McGlincy, N.J., & Ingolia, N.T. (2017). Transcriptome-wide measurement of translation by ribosome profiling. Methods, 126, 112-129.
| ||

| MG | RNA-seq - Préparation de librairie | |
|---|---|---|
RNA-seq - Préparation de librairie I) Objectifs Le séquençage d'ARN (RNA-Seq) utilise les capacités des méthodes de séquençage à haut débit pour fournir un aperçu du transcriptome d'une cellule. Par rapport aux méthodes précédentes de Sanger (1) et de microarray (2), le RNA-Seq offre une couverture beaucoup plus élevée et une plus grande résolution de la nature dynamique du transcriptome. Au-delà de la quantification de l'expression des gènes, les données générées par le RNA-Seq facilitent la découverte de nouveaux transcrits, l'identification de transcrits issus d’épissage alternatif et la détection de l'expression spécifique des allèles. Les améliorations récentes du workflow du RNA-Seq, de la préparation de l'échantillon à la construction de la bibliothèque jusqu'à l'analyse des données, ont permis aux chercheurs d'élucider davantage la complexité fonctionnelle de la transcription. En plus des transcrits d'ARN messager (ARNm) polyadénylés, le RNA-Seq peut être appliqué pour étudier différentes populations d'ARN, y compris l'ARN total, les pré-ARNm, et les ARN non codant, tels que les microARN et les ARNc long. (3)
II) Principe La bibliothèque d'ADNc est générée par transcription inverse comprenant des adaptateurs de séquençage spécifiques avec des séquences « codes-barres ». Ensuite, les bibliothèques sont regroupées sur des plaques, les flowcells, et séquencées sur différentes machines (ex : Illumina). Le nombre envisagé de lectures par bibliothèque dépend de l'organisme étudié et de la sensibilité souhaitée. Alors que la référence pour les génomes eucaryotes complexes (par exemple humain, rat, souris) nécessite des lectures de 100-150 nt (sensibilité élevée) ou 20-30 nt lectures (faible sensibilité), une quantité de lecture 10 fois plus faible est requise pour les bactéries. (4)
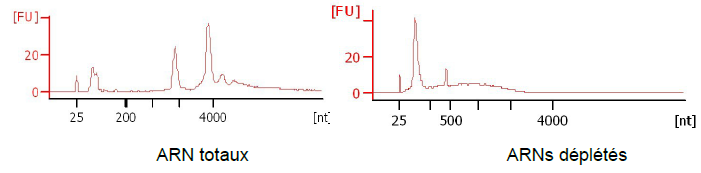
III) Mode opératoire Une extraction des ARN totaux est réalisée. Ces ARN sont ensuite contrôlés sur Bioanalyser (Agilent) pour vérifier leur intégrité (figure 1).
Figure 1 : Comparaison des courbes d’ARN total et d’ARN déplétés (4) Les ARNm matures sont ensuite extraits au moyen de billes portant des séquences polyT auxquelles les queues polyA, spécifiques des ARNm matures, se lient (figure 2).
Figure 2 : Purification des ARNm matures (5) Les ARNm sont transcrits en ADNc double brin par réverse transcription, et ceux-ci sont fragmentés chimiquement en fragments d’environ 200 pb. Enfin, on lie des adaptateurs aux deux extrémités des fragments d’ADNc double brin. Ces adaptateurs sont composés de séquences orientées et spécifiques : - à l’échantillon (séquence « Ech ») : ce sont des séquences codes-barres nécessaires au multiplexage (car elles permettent de distinguer plusieurs échantillons sur une même flowcell) - à la flowcell (séquences P1 et P2) : elles correspondent aux cotés 3’ et 5’ et peuvent être utilisées pour une pré-amplification PCR. Elles sont compatibles avec la flowcell où sont fixés des oligonucléotides permettant le séquençage. (6)
Figure 3 : Organisation des ADNc finaux (7)
Figure 4 : Workflow de la préparation d’une librairie d’ADNc pour le RNA-seq (8) IV) Présentation des résultats A l’issu de la préparation de librairie une solution contenant les fragments d’ADNc à séquencer est obtenue. Ces fragments sont ensuite déposés sur une plaque sur laquelle sont fixés des oligonucléotides correspondants aux séquences P1 et P2 (voir partie mode opératoire). Cette plaque ou flowcell est ensuite placée dans un séquenceur (ex : séquenceur Illumina).
V) Interprétation des résultats La préparation de librairie en elle-même ne fournit aucun résultat. Le séquençage des librairies permet la découverte de nouveaux gènes ou de déterminer le niveau d’expression d’un gène donné.
VI) Intérêts et limites Un intérêt majeur du RNA-seq est la possibilité d’un multiplexage, à savoir une analyse simultanée de plusieurs échantillons grâce aux séquences barcode. Certaines manipulations au cours de la construction de la bibliothèque compliquent l'analyse des résultats de RNA-seq. Par exemple, de nombreuses « courtes lectures » peuvent être obtenues. Celles-ci sont identiques les unes aux autres, à partir des banques d'ADNc qui ont été amplifiées. Celles-ci pourraient être un véritable reflet des espèces d'ARN abondantes, ou ils pourraient être des artefacts de PCR. Une façon d’identifier l’origine de ces lectures courtes est de déterminer si les mêmes séquences sont observées dans différentes répliques biologiques. Une autre considération importante concernant la construction de la bibliothèque est de savoir s'il faut ou non préparer des bibliothèques spécifiques aux brins. Ces bibliothèques ont l'avantage de fournir des informations sur l'orientation des transcrits, ce qui est très utile pour l'annotation du transcriptome, en particulier pour les régions ayant des transcriptions chevauchantes et de directions opposées. Cependant, les bibliothèques spécifiques aux brins sont laborieuses à produire parce qu'elles nécessitent de nombreuses étapes ou une ligation ARN-ARN directe, qui est peu efficace. En outre, il est essentiel de s'assurer que les transcrits anti-sens ne sont pas des artefacts de transcription inverse. En raison de ces complications, la plupart des études jusqu'à présent ont analysé des ADNc sans information de brin. (9)
Références bibliographiques
1. Thermofisher. A simplified DNA extraction method for Sanger sequencing of FFPE samples. [en ligne]. Disponible à l’adresse : http://tools.thermofisher.com/content/sfs/brochures/dna-extraction-sanger-sequencing-FFPE-app-note.pdf 2. ROGLER, C. E., TCHAIKOVSKAYA, Tatyana, NOREL, Raquel, MASSIMI, Aldo, PLESCIA, Christopher, RUBASHEVSKY, Eugeny, SIEBERT, Paul et ROGLER, Leslie E. RNA expression microarrays (REMs), a high-throughput method to measure differences in gene expression in diverse biological samples. Nucleic Acids Research. 2004. Vol. 32, n° 15, pp. e120. DOI 10.1093/nar/gnh116. PMID: 15329382PMCID: PMC516075 3. KUKURBA, KR et MONTGOMERY. An introduction to RNA-Seq methods, applications, experimental design, and technical challenges | RNA-Seq Blog. RNA-Seqblog [en ligne]. Disponible à l’adresse : http://www.rna-seqblog.com/an-introduction-to-rna-seq-methods-applications-experimental-design-and-technical-challenges/ 4. DUBOIS, Emeric. Fiche prestation : Séquençage RNA-Seq - Documentation - Montpellier GenomiX. [en ligne]. 4 mars 2015. Disponible à l’adresse : https://www.mgx.cnrs.fr/project/documents/537 5. BOUCHEZ, Olivier et MARSAUD, Nathalie. RNA-seq. Genotoul [en ligne]. 28 mars 2012. Disponible à l’adresse : http://get.genotoul.fr/fileadmin/user_upload/Presentation/PlaGe/120328_animation_RNAseq.pdf 6. MICROSYNTH. RNA Sequencing For Differential Gene Expression Analysis. [en ligne]. [Consulté le 4 mai 2017]. Disponible à l’adresse : http://www.microsynth.ch/index_de.php?TPL=10607&gclid=CjwKEAjwtbPGBRDhoLaqn6HknWsSJABR-o5sWrom5RRQDtiCOzH3rTbjcnBXKzrRBNlvDTPhunIDshoCuCzw_wcB 7. LAGARRIGUE, Sandrine. (2017). Cours Transcriptomique, UE Génétique et génomique. AGROCAMPUS OUEST. 8. BLERVAQUE, Renaud. Survol de la technologie de séquençage PGM Ion Torrent. Biorigami [en ligne]. Disponible à l’adresse : http://www.biorigami.com/?tag=principe-ion-torrent-pgm 9. WANG, Zhong, GERSTEIN, Mark et SNYDER, Michael. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews. Genetics. janvier 2009. Vol. 10, n° 1, pp. 57‑63. DOI 10.1038/nrg2484. PMID: 19015660PMCID: PMC2949280 | ||



| MS | RNAscope | ||
|---|---|---|---|
RNAscope I) ObjectifsLe RNAscope est une méthode d’hybridation in situ permettant de visualiser, localiser et quantifier spatialement l’expression d’ARN cibles au niveau cellulaire, tout en préservant la structure tissulaire et en supprimant le bruit de fond. La méthode du RNAscope est actuellement la seule permettant de détecter et visualiser des ARN non codant dans le contexte cellulaire. II) PrincipeLa méthode du RNAscope permet de visualiser la présence de n’importe quel type d’ARN dans n’importe quel tissu de n’importe quelle espèce (ou du moins, c’est ce qu’annonce ACD, l’entreprise qui détient le RNAscope). A l’aide de sondes fluorescentes préalablement conçues, cette détection peut se faire sur tout type d’ARN (même sur celui d’espèces peu étudiées). Quelques exemples de molécules détectables par cette méthode : ARN très faiblement exprimés, nouveaux isolats de virus, ARN non-codant, ARN codant pour des récepteurs pour lesquels il n’existe pas de bons anticorps… (Advanced Cell Diagnostics, 2014a). La quantification d’ARN par le RNAscope repose donc sur l’hybridation de cette molécule avec plusieurs paires de sondes spécifiques. Des marqueurs fluorescents se fixent ensuite à ces sondes, ce qui permet l’amplification du signal et la facilitation de l’observation de l’ARN.
III) Mode opératoirePréparation des échantillons et hybridation Les échantillons subissent tout d’abord un traitement spécifique engendrant leur perméabilisation : cette étape est nécessaire afin de permettre l’hybridation des ARN aux sondes. Une sonde à ARN ainsi que des protéases sont ajoutées aux tissus ou cellules à étudier (Advanced Cell Diagnostics, 2014a). Ces sondes, appelées sondes cibles Z, ont une partie “inférieure” complémentaire de la séquence des ARN dont la présence est à tester, et une partie “supérieure” constituée d’une séquence de 14 nucléotides. Les régions inférieures et supérieures sont séparées par la séquence d’espacement. Ces sondes forment des paires, dont les parties supérieures libres forment un site de fixation pour le préamplificateur de 28 bases (Advanced Cell Diagnostics, 2015). Amplification Seules les paires de sondes cibles Z seront reconnues par le préamplificateur, qui se fixe sur les deux régions supérieures adjacentes. Ce phénomène permet une certaine spécificité, puisque les sondes non appariées ne permettront pas au préamplificateur de se lier de façon suffisamment forte pour supporter la phase de lavage (Advanced Cell Diagnostics, 2014). Une fois le préamplificateur fixé à la paire de sondes cibles Z, les amplificateurs peuvent se lier aux sites spécifiques de celui-ci. Enfin, des sondes marquées de fluorescence (molécules de fluorescence ou enzymes chromogéniques) se fixent sur les amplificateurs (Advanced Cell Diagnostics, 2015). Visualisation Un signal est visible dès que 3 couples de sondes cibles Z sont fixés à l’ARN cible. La méthode RNAscope produit cependant 20 couples de sondes cibles Z, afin d’augmenter la puissance du signal observé. La grande spécificité de ces sondes permet la distinction de différents sous-types de molécules (Advanced Cell Diagnostics, 2015). Quantification La quantification des ARN se fait soit manuellement avec le kit fourni par ACD, soit automatiquement avec le logiciel RNAscope SpotStudio Software. L’évaluation manuelle du nombre d’ARN est semi-quantitative. Lors de l'interprétation de la coloration RNAscope, il est recommandé de noter le nombre de points par cellule plutôt que l'intensité du signal, car le nombre de points est corrélé au nombre de copies d'ARN, alors que l'intensité des points reflète le nombre de paires de sondes liées à chaque molécule. Lorsqu’une quantification automatique est effectuée, une image du tissu est prise (par exemple par caméra microscopique), puis chargée sur le logiciel. L’utilisateur règle certains paramètres (identification de la cellule, diamètre du noyau, diamètre des points représentant les ARN, …). Le logiciel renvoie alors une image avec les cellules délimitées selon un code couleur correspondant au nombre de molécules d’ARN présentes dans la cellule, et un tableau de données avec le nombre d’ARN par cellule pour permettre un traitement statistique des données (Advanced Cell Diagnostics, 2014b).
Figure 2 : Analyse de coupe de tissu avec le logiciel RNAscope SpotStudio Software (Advanced Cell Diagnostics, 2014a) IV) Présentation des résultatsDans cet exemple, des coupes de foie de souris injectées à la Concanavaline A (modélisation d’une hépatite auto-immune) ont été traitées grâce à la technique du RNAscope. Deux sondes ont été utilisées sur chaque coupe. Le but de la manipulation était d’identifier les cellules productrices d’IL-18 dans le foie dans un modèle d’hépatite aiguë de cause auto-immune.
Figure 3 : Coupe de foie murin traité à la ConA, sonde A (fluorochromes bleu) : détection ARNm PECAM 1 (spécifique des cellules endothéliales), sonde B (fluorochromes rouges) : détection ARNm IL-18 (molécule d’intérêt) (Stosskopf, 2022)
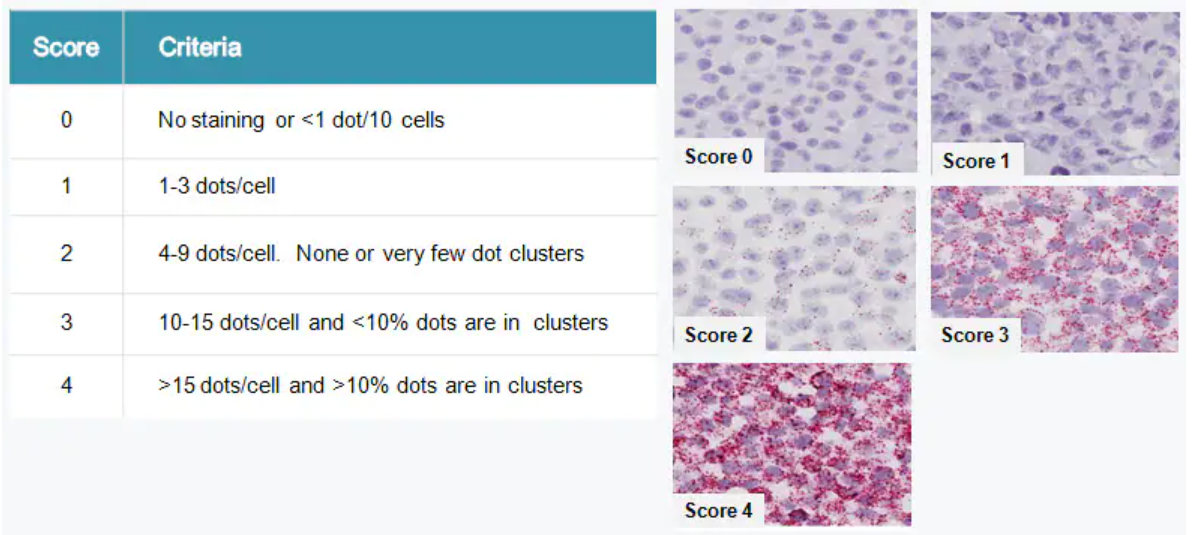
Figure 4 : Coupe de foie murin traité à la ConA, sonde A (fluorochromes bleu) : détection ARNm Kératine 18 (spécifique des hépatocytes), sonde B (fluorochromes rouges) : détection ARNm IL-18 (molécule d’intérêt) G : lumière blanche, D : même image en lumière fluorescente, canal TRITC. (Stosskopf, 2022) V) Interprétation des résultats(Lors de cette manipulation, nous n’avons pas eu accès au logiciel de quantification, donc seule la méthode manuelle semi-quantitative sera décrite ici.) Localisation : Sur la figure 3, les fluorochromes bleus (marquant les ARNm PECAM 1) et rouges (marquant les ARNm IL-18) ne sont pas colocalisés : les cellules endothéliales ne sont donc pas les cellules sources d’IL-18 dans le foie dans le modèle de la ConA. Sur la figure 4, il apparaît une colocalisation des fluorochromes bleus (marquant les ARNm Kératine 18) et rouges (marquant les ARNm IL-18) : les hépatocytes sont donc responsables de la production d’IL-18 dans le foie dans le modèle d’hépatite induite par la ConA. Plus généralement, la visualisation des tissus colorés grâce à la technique du RNAscope permet de localiser clairement un ARN d’intérêt, qu’il soit codant ou non. Plus précisément, l’utilisation de 2 sondes (test RNAscope en duplex) permet de localiser les cellules sources d’un ARNm d’intérêt. Quantification : Grâce à la grille de score ci-dessous fournie par le producteur du kit, il est possible d’évaluer semi-quantitativement et manuellement les ARNm présents dans les deux coupes de foie (Figures 3 et 4). L’ARNm PECAM 1 obtient un score de 3, l’ARNm Kératine 18 a un score de 4 et l’ARNm IL-18 obtient un score de 2. L’absence de témoin négatif rend ces résultats difficilement interprétables.
Figure 5 : Interprétation des résultats : quantification (manuelle) des ARN en fonction de la coloration (staining) de l’échantillon (Advanced Cell Diagnostics, 2015) VII) Intérêts et limitesCette méthode présente de nombreux intérêts : elle permet notamment la localisation et quantification de plusieurs ARN (quatre maximum) simultanément, même si l’ARN d’intérêt est faiblement exprimé dans le tissu. La technique du RNAscope étant une méthode très reproductible, elle permet des résultats plus fiables, et disponibles en 8 heures. Cependant, cette méthode reste coûteuse, et la quantification manuelle peut être laborieuse et peu précise (dépendante de l’observateur). De plus, une utilisation de cette méthode in vivo est très limitée, voire impossible, puisqu’il faut nécessairement prélever un tissu d’intérêt à observer. VII) Références bibliographiquesAdvanced Cell Diagnostics (2014a) RNAscope® - a novel breakthrough RNA in situ hybridization platform. Advanced Cell Diagnostics (2015) ACD RNAscope in situ Hybridization (ISH) Technology Overview. Advanced Cell Diagnostics (2014b) In Situ Hybridization Kits, Hybridization Assays | Manual Assays. https://acdbio.com/manual-assays-rnascope Avanced Cell Diagnostics RNAscope Assay Technical Support and FAQ | Support. https://acdbio.com/technical-support/getting-started Stosskopf M. (2022) Rapport de stage Mission | Étude du rôle d’IL-18 BP au cours de l’hépatite aiguë induite par la ConA chez la souris.
| |||