Wiki des méthodes de biologie moléculaire et cellulaire
Glossaire collaboratif des méthodes expérimentales de biologie moléculaire et cellulaire. Ce glossaire est réalisé par les étudiants du module "Méthodes expérimentales de biologie moléculaire et cellulaire" sous la supervision de l'équipe de Génétique Animale.
Spécial | A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z | Tout
F |
|---|


| MP | FISH (Fluorescence In Situ Hybridization) | |
|---|---|---|
I) Objectifs
La méthode FISH est une méthode de cytogénétique permettant de détecter et de localiser des séquences d'ADN spécifiques sur un chromosome, des séquences d'ARN ou encore des protéines, directement au sein des cellules ou tissus étudiés. Les sondes utilisées sur l'ADN peuvent cibler les chromosome entier, le centromère, ou encre un locus particulier. Cette méthode permet de faciliter le caryotypage, et est utilisée en médecine pour la détection de maladies génétiques, et en sélection, pour identifier les gamètes déséquilibrées. Elle peut également permettre de différencier les espèces, et donc identifier une bactérie à l'aide de son ADN.
II) Principe
L'hybridation in situ en fluorescence se réalise en mettant en contact des sondes spécifique aux séquences recherchées avec la séquence à tester. Une fois hybridées, les sondes sont repérées à l'aide d'un marqueur fluorescent, visible à l'aide d'un microscope adapté.
III) Mode opératoire
Tout d'abord, une sonde est préparée par constitution d'un brin d'ADN comportant des nucléotides modifiés. Elle doit être assez longue pour pouvoir s'hybrider avec la zone de l'ADN ou de l'ARN voulue, mais pas trop grande pour ne pas être un frein à l'hybridation. Les sondes peuvent être directement étiquetées par des fluorochromes, ou repérées plus tard par fixation d'anticorps flanqués de molécules fluorescentes ou encore à l'aide de la biotine. Les cellules que l'on veut analyser sont ensuite fixées, puis mises en présence des sondes. L'ADN et les sondes sont dénaturés par chauffage, puis, lorsque la température redescend, ils s'hybrident. La solution est ensuite lavée plusieurs fois pour éliminer les sondes non hybridées. Si la reconnaissance des sondes se fait à l'aide d'anticorps, on mets alors les anticorps, qui se fixent aux sondes hybridées, et on effectue de nouveau des lavages pour éliminer le surplus d'anticorps. On peut ensuite observer les cellules à l'aide d'un microscope à épifluorescence, qui excite les molécules fluorescentes, qui sont émettent alors une lumière, visible au microscope. Il existe différentes molécules fluorescentes, et on peut donc repérer plusieurs sites en même temps, en utilisant des couleurs différentes par exemple.
IV) Présentation des résultats
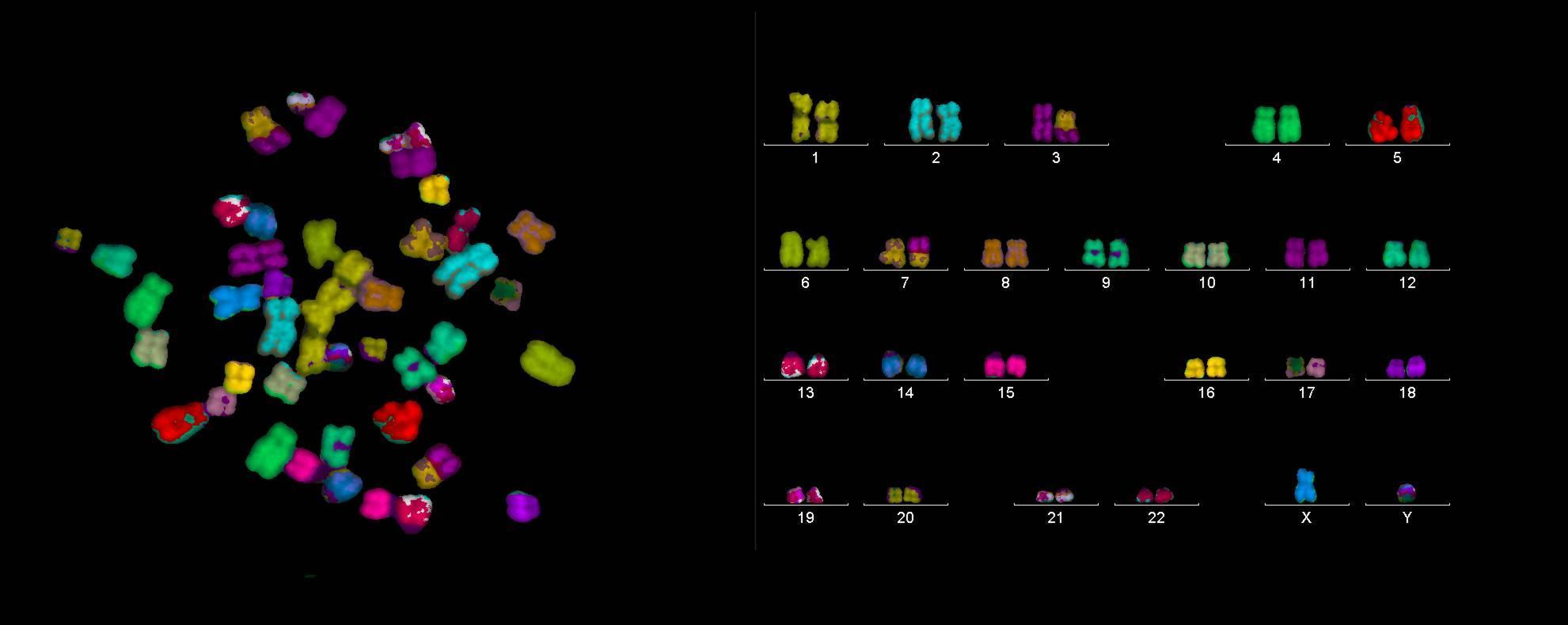
Les résultats sont très visuels : ils consistent en une photographie (ou une vidéo si la cellule n'est pas fixée) sur laquelle on peut voir de différentes couleurs les zones d'intérêt. Lorsque la méthode est utilisée pour aider au caryotypage, on peut ajouter le caryotype en parallèle de la photo prise au microscope. Figure 2 : Caryotypage d'un humain pour recherche des aberrations chromosomiques (https://www.nasa.gov/mission_pages/station/research/experiments/271.html)
V) Interprétation des résultats
Parfois, l'interprétation est très simple : lorsqu’il s'agit simplement de détecter la présence ou non d'une molécule, il suffit de regarder s'il y a fluorescence ou non. Sinon, il faut analyser la photo obtenue : on peut pas exemple compter le nombre de chromosomes, repérer des translocations en remarquant une alternance de couleurs au sein d'un même chromosome, identifier les zones de chromatine, etc … Enfin des sondes spécifiques de locus peuvent être utilisées, et dans ce cas on peut déterminer s'il y a eu une mutation par délétion, lorsque la zone attendue ne présente pas de fluorescence.
VI) Intérêts et limites
L'avantage de cette méthode est qu'elle peut à la fois être réalisée sur des cellules fixées ou sur des cellules vivantes. De plus, la structure et l'organisation de la chromatine ne sont pas perturbées. Cette méthode permet la visualisation directe des chromosomes lors de l'interphase.
Le problème est que suite au traitement de la FISH, la cellule n'est plus viable. De plus, la fluorescence reste limitée dans le temps et l'analyse doit être faite rapidement, même si ce problème peut être contré par la prise de photo. L'analyse de résultats en deux dimensions peut limiter la vision et altérer les résultats. Enfin, cette technique reste chère, car il faut se procurer les sondes spécifiques aux régions à tester.
VII) Références bibliographiquesPolak, Julia M. and James O’D McGee. 1990. In Situ Hybridization: Principles and Practice. Oxford University Press, Oxford, pp. 10-30.
Schrock, E., et al. 1996. Multicolor spectral karyotyping of human chromosomes. Science 273, 494–497 doi:10.1126/science.273.5274.494
Dohm, Juliane C., et al. 2013. The genome of the recently domesticated crop plant sugar beet (Beta vulgaris). Nature 505 (7484): 546‑49. doi:10.1038/nature12817. | ||

| PB | Förster Resonance Energy Transfert | |
|---|---|---|
FRET (Förster Resonance Energy Transfert)
I) Objectifs
Le FRET est un processus par lequel de l’énergie d’un fluorophore donneur à l’état excité est transmis à fluorophore accepteur situé à proximité immédiate (distance <10nm). Cela permet donc de détecter les interactions intramoléculaires, telles qu’une interaction protéine/ protéine ou les changements de conformation. Il est alors possible de savoir où et quand, dans une cellule, des molécules d’intérêt interagissent.
II) Principe
Cette technique utilise deux molécules fluorescentes, chacune rattachée à une molécule d’étude (les deux fluorophores pouvant être sur la même molécule). L’un des deux est un fluorophore donneur et l’autre est accepteur. Le principe est d’exciter le fluorophore donneur. Lors de la lecture des résultats, si la fluorescence majoritaire correspond au donneur, c’est qu’il n’y a pas eu de transfert d’énergie et donc pas d’interaction. Inversement, si la fluorescence majoritaire vient du receveur, il y a eu un transfert d’énergie et donc une interaction entre les molécules étudiées. Il est aussi possible de ne mesurer que l’émission du donneur et le temps de celle-ci. Si le temps d’émission est cours, il y a interaction.
III) Mode opératoire
- La première étape de la méthode consiste à savoir quelle stratégie utiliser. La fluorescence peut être à l’état naturel dans la cellule ou alors être ajoutée de manière artificielle. Si le fluorophore est ajouté, celui-ci peut être lié à un constituant cellulaire d’étude, à un anticorps d’une molécule cible ou encore via l’ajout d’un ADNc codant pour un fluorophore à l’ADNc codant pour la protéine d’intérêt. En connaissant cela, il est alors possible de choisir une stratégie, soit utiliser deux protéines couplées aux fluorophores, soit une protéine et un anticorps ciblé contre une autre protéine, protéine et anticorps couplés à un fluorophore. - Il faut ensuite définir la place de l’étiquette fluorescente, d’après les caractéristiques connues des protéines d’intérêt, telles que les propriétés biologiques, les données structurales. - Un fois l’étiquette placée, il est nécessaire de tester l’ajout de celle-ci sur les propriétés des protéines d’intérêt, par rapport aux endogènes, comme la localisation subcellulaire ou les propriétés enzymatiques le cas échéant.
- Il est ensuite possible d’étudier la colocalisation des protéines d’intérêt, via la microscopie confocale, ou alors leurs interactions.
IV) Présentation des résultats
Selon le type d’analyse que l’utilisateur souhaite, les résultats peuvent se présenter sous plusieurs formes. Si l’utilisateur souhaite voir la localisation d’interaction déjà connue, les résultats peuvent être présentés sous la forme de photographies prises au microscope à fluorescence. Les fluorescences peuvent correspondre au temps de vie du fluorophore donneur ou alors à la fluorescence du fluorophore accepteur. L’utilisateur peut aussi chercher à montrer qu’il y a une interaction entre deux composants d’une cellule. Les résultats se présentent alors sous la forme de graphiques indiquant la durée de vie du fluorophore donneur.
V) Interprétation des résultats
Dans le cadre de la recherche d’interaction, la mesure de la durée de vie du fluorophore donneur se fait dans deux conditions. La première correspond au témoin, c’est-à-dire le fluorophore donneur seul et constitue donc une durée vie de référence. La seconde correspond aux conditions test, c’est-à-dire que les fluorophores accepteur et donneur sont présents dans la cellule. S’il y a interaction entre les molécules d’étude, l’émission du donneur sera absorbée par l’accepteur. La durée de vie du donneur sera alors diminuée par rapport au témoin. Pour la localisation, l’utilisateur a juste besoin d’analyser les images issues de la microscopie, les points de fluorescence indiquent la localisation des molécules d’étude (intra-membranaire, cytoplasmique …) et l’intensité indique la proportion de molécules à ces endroits.
VI) Intérêts et limites
Cette technique a pour intérêt de savoir où et quand dans une cellule des molécules d’intérêt interagissent ; ce que d’autres techniques expérimentales ne permettent pas d’obtenir. Par contre, dans cette technique, des résultats négatifs ne permettent pas d’affirmer que les molécules d’intérêt n’interagissent pas. De plus, cette technique suppose un niveau d’expression élevé des molécules d’intérêt, ce qui peut perturber leur fonction et/ ou la réponse cellulaire.
VII) Références bibliographiques
Clapp, Aaron R., Igor L. Medintz, and Hedi Mattoussi. 2006. “Förster Resonance Energy Transfer Investigations Using Quantum-Dot Fluorophores.” ChemPhysChem 7 (1): 47–57. doi:10.1002/cphc.200500217. Clegg, Robert M. 1995. “Fluorescence Resonance Energy Transfer.” Current Opinion in Biotechnology 6 (1): 103–10. doi:10.1016/0958-1669(95)80016-6.
| ||
| RH | FUCCI | ||
|---|---|---|---|
La méthode FUCCI, mise au point en 2008 (Sakaue-Sawano et al., 2008), aussi appelée indicateur de cycle cellulaire basé sur l’ubiquitination fluorescente (Fluorescence Ubiquitination Cell Cycle Indicator) est une méthode permettant de distinguer la phase G1 d’une cellule des phases S/G2/M (Yano et al., 2020). C’est une méthode qui permet l’observation dans l’espace et le temps des cellules grâce à un système de fluorescence. La couleur de la fluorescence dépend de la phase dans laquelle se trouve la cellule. Ainsi, les cellules apparaissent rouges/oranges lors de la phase G1 et vertes lors des phases S/G2/M (Zielke and Edgar, 2015). Cet outil est particulièrement utile dans la recherche en biologie cellulaire, permettant aux chercheurs d'étudier la dynamique du cycle cellulaire et la prolifération dans divers types de cellules, y compris les cellules cancéreuses. FUCCI utilise des protéines naturellement présentes et essentielles dans le cycle cellulaire. Ainsi, les sondes fluorescentes rouges/oranges et vertes sont liées à des fragments de ces protéines, respectivement Cdt1 s’exprimant au cours de la phase G1 et la géminine s’exprimant durant les phases S/G2/M. (Tada, 2007) Lors de la transition entre la phase G1 et la phase S, pendant une courte durée, les deux sondes sont présentes et s’expriment, ce qui laisse apparaître une couleur jaune de façon très succincte (Zielke and Edgar, 2015). Le système FUCCI se compose de deux sondes fluorescentes construites à partir de protéines fluorescentes de différentes couleurs, chacune fusionnée à un régulateur du cycle cellulaire : Cdt1 et la géminine (Zielke and Edgar, 2015). Cdt1(Cdc10 dependent transcript 1) est un facteur de réplication se fixant sur le complexe ORC (Origin Recognition Complex), lui-même fixé sur l’ADN, durant la phase G1 du cycle cellulaire. Lorsque la phase S débute et que la réplication commence, une interaction entre Cdt1 et la géminine entraîne l’inactivation de Cdt1, qui sera par la suite dégradé par protéolyse. Pour ne pas interférer avec Cdt1 et lui permettre d’assurer son rôle, la géminine est inactive lors de la phase G1. Ainsi, Cdt1 s’exprime durant la phase G1, alors que la géminine est fonctionnelle durant les phases S/G2/M du cycle cellulaire (Blanchard, 2003). La Figure 1 illustre ces mécanismes. Pour élaborer les sondes fluorescentes du système FUCCI, deux protéines fluorescentes sont utilisées : mKO2 (monomère Kusabira-Orange 2) et mAG1 (monomère Azami Green). mKO2 est fusionné à un fragment humain de la protéine Cdt1 (acides aminés 30 à 120), tandis que mAG1 est fusionné à un fragment humain de la géminine (acides aminés 1 à 110). Ainsi, lorsque les protéines Cdt1 seront fonctionnelles et actives, elles émettront respectivement une fluorescence rouge pendant la phase G1 ou une fluorescence verte pendant les phases S/G2/M. Les fragments de protéines humaines ont été choisis de sorte à ce que les sites actifs soient conservés et que le cycle cellulaire ne soit pas perturbé (Zielke and Edgar, 2015). Les sondes sont schématisées en Figure 2. Les sondes peuvent ensuite être insérées dans les noyaux des cellules grâce à des vecteurs comme les lentivirus. C’est une technique très efficace qui a déjà permis d’obtenir des cellules exprimant les deux sondes à des niveaux équivalents (Sakaue-Sawano et al., 2008).
Figure 2. Composition des sondes fluorescentes utilisées dans la méthode FUCCI (Sakaue-Sawano et al., 2008) Ainsi, la fluorescence des cellules change au cours du cycle cellulaire, ce qui permet de visualiser les différentes phases du cycle cellulaire. De plus, lors du passage de la phase G1 à la phase S, les deux sondes sont exprimées simultanément pendant une courte période, ce qui entraine momentanément l’apparition d’une couleur jaune. La figure 3 représente les couleurs et les sondes exprimées au cours du cycle cellulaire.
Figure 3. (a) Couleurs observées durant le cycle d’une cellule (Kaida and Miura, 2021) (b) Variation de la fluorescence des deux sondes au cours du cycle cellulaire (Zielke and Edgar, 2015) IV) Présentation des résultats Pour connaître le stade du cycle cellulaire à un instant T de chaque cellule présente dans une culture, il est possible d’avoir recours à la cytométrie de flux. La protéine mAG1 est excitée par des longueurs d’onde autour de 492 nm et émet à 505 nm. Quant à mKO2, l’excitation se fait autour de 551 nm et l’émission autour de 565 nm. Grâce à ces longueurs d’onde, la cytométrie de flux permet de visualiser la quantité de cellule en phase G1, en phase S/G2/M ou même en transition entre les phases G1 et S. Un exemple de résultat obtenu par cytométrie de flux est représenté en Figure 4. Figure 4. Résultats de la méthode FUCCI observés par cytométrie de flux (Sakaue-Sawano et al., 2008) En revanche, pour observer l’évolution du cycle cellulaire d’une ou plusieurs cellules en temps réel, l’utilisation d’un microscope à fluorescence sera plus adaptée pour permettre la visualisation continue des cellules en temps réel. Un exemple de résultat obtenu par microscopie à fluorescence est montré en Figure 5. Figure 5. Résultats de la méthode FUCCI observés par microscopie à fluorescence (Sakaue-Sawano et al., 2008) V) Interprétation des résultats L’interprétation des résultats sera permise par la couleur observée : une cellule exprimant une fluorescence rouge/orange est en phase G1 tandis qu’une fluorescence verte est significative des stades G2, S ou M. De plus, si l’on arrive à observer une fluorescence jaune, c’est que la cellule se trouve en phase de transition entre les phases G1 et S du cycle cellulaire. L'utilisation de ces résultats et leur interprétation dépendent de la question de recherche abordée. Par exemple, si l'objectif est d'étudier l'effet d'un médicament sur la progression du cycle cellulaire, les résultats pourraient montrer des changements dans la durée de chaque phase du cycle cellulaire. En revanche, si l'objectif est d'étudier le cycle cellulaire des cellules cancéreuses, les résultats permettraient d’identifier une progression anormale du cycle cellulaire, qui pourrait être utilisée comme cible pour une thérapie anticancéreuse. L'utilisation de la méthode FUCCI présente plusieurs avantages. Il s'agit d'une méthode non invasive pour l'imagerie des cellules vivantes, qui permet de visualiser les changements dynamiques dans la progression du cycle cellulaire. En outre, elle permet d'identifier les anomalies du cycle cellulaire, telles qu’un arrêt ou une accélération. Cela peut aider à comprendre les mécanismes sous-jacents de maladies telles que le cancer. L'une des limites de la FUCCI est qu'elle ne fournit que des informations sur les phases du cycle cellulaire. Elle ne fournit aucune information sur les points de contrôle du cycle cellulaire. De plus, il peut être difficile de distinguer les phases S et G2 en utilisant la méthode FUCCI, car la fluorescence mAG1 est présente dans les deux phases. Une amélioration de la méthode FUCCI a été élaborée pour surpasser cette limite en utilisant des sondes de couleurs différentes liées à des protéines spécifiques de chacune de ces deux phases. VII) Références bibliographiques Blanchard J-M (2003) Des oncogènes aux régulateurs de la mitose : un changement de perspective dans notre vision des processus cancéreux. Med Sci (Paris) 19: 187–199 Kaida A, Miura M (2021) Visualization of Radiation-Induced Cell Cycle Kinetics with a Fluorescent Ubiquitination-Based Cell Cycle Indicator (Fucci). In AS Coutts, L Weston, eds, Cell Cycle Oscillators. Springer US, New York, NY, pp 223–236 Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al (2008) Visualizing Spatiotemporal Dynamics of Multicellular Cell-Cycle Progression. Cell 132: 487–498 Tada S (2007) Cdt1 and geminin: role during cell cycle progression and DNA damage in higher eukaryotes. Front Biosci 12: 1629 Yano S, Tazawa H, Kagawa S, Fujiwara T, Hoffman RM (2020) FUCCI Real-Time Cell-Cycle Imaging as a Guide for Designing Improved Cancer Therapy: A Review of Innovative Strategies to Target Quiescent Chemo-Resistant Cancer Cells. Cancers 12: 2655 Zielke N, Edgar BA (2015) FUCCI sensors: powerful new tools for analysis of cell proliferation: FUCCI sensors. WIREs Dev Biol 4: 469–487 Réalisé par Roxane HERTZ & Charline BATEL, 2023 | |||




