Wiki des méthodes de biologie moléculaire et cellulaire
Glossaire collaboratif des méthodes expérimentales de biologie moléculaire et cellulaire. Ce glossaire est réalisé par les étudiants du module "Méthodes expérimentales de biologie moléculaire et cellulaire" sous la supervision de l'équipe de Génétique Animale.
Spécial | A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z | Tout
S |
|---|
| CD | SEQUENÇAGE AUTOMATISE SELON LA METHODE DE SANGER | |
|---|---|---|
Séquençage automatisé selon la méthode de Sanger
Objectifs Cette méthode, aussi appelée “dideoxynucleotide sequencing” 1 ou “chain termination sequencing”1, a pour but de séquencer des échantillons d’ADN, donc de connaître l’ordre dans lequel sont positionnés les 4 nucléotides : A (Adénine), C (Cytosine), G (Guanine) et T (Thymine).
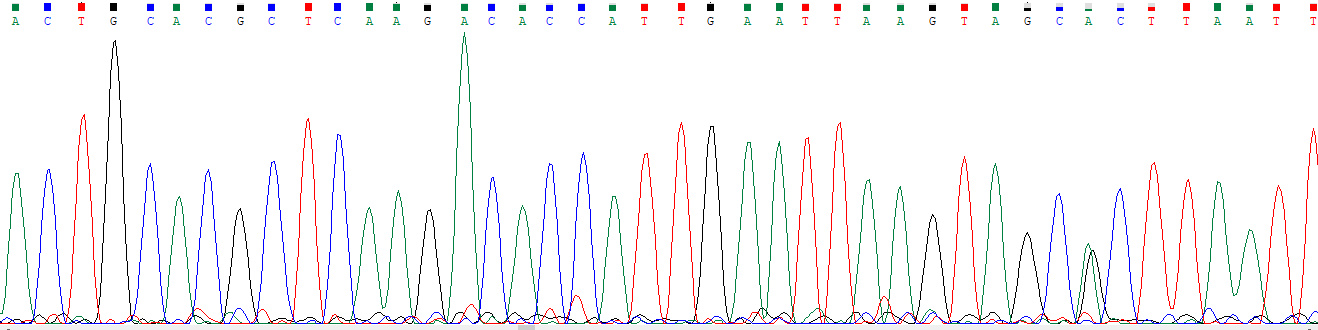
Principe (Figure 1) Le principe se base sur l’ADN polymérase et les di-déoxyribonucléotides (ddNTP). Les échantillons d’ADN à séquencer sont mélangés avec des amorces spécifiques, de l’ADN polymérase, des déoxyribonucléotides (dATP, dCTP, dGTP et dTTP) et des di-déoxyribonucléotides (sans groupement hydroxyle en position 3’) (ddATP, ddCTP, ddGTP et ddTTP). L’incorporation de ces ddNTP empêche la formation de la liaison phospho-diester, et donc la poursuite de l’élongation du brin d’ADN². Des fragments d’ADN de tailles variables se terminant par un ddNTP sont ainsi obtenus. Dans la première version de cette méthode3, les amorces étaient marquées radioactivement et les bouts d’ADN migraient sur un gel dénaturant permettant l’obtention d’un profil de migration3. Actuellement, une amplification par PCR est réalisée en amont, et la méthode employée utilise l’électrophorèse capillaire et la fluorescence. L’électrophorèse permet de classer les fragments selon leur taille et ainsi obtenir la séquence nucléotidique. De plus, chaque ddNTP est marqué par un fluorochrome différent. Un rayonnement laser excite la molécule fluorescente qui réémet une longueur d’onde spécifique. Cette dernière est détectée par une caméra. A la fin du séquençage, cela permet d’obtenir un électrophorégramme.
Figure 1 : Principe du séquençage Sanger 4
Figure 2 : Exemple de résultats du séquençage sur Chromas, communication personnelle
Figure 3 : Options d’utilisation de la méthode Sanger ou NGS 6 4-Khan Academy (modifié en 2020), Dna sequencing, Method of Sanger sequencing, Khan Academy (en ligne). https://www.khanacademy.org/science/high-school-biology/hs-molecular-genetics/hs-biotechnology/a/dna-sequencing (consulté le 18/03/2020) 5- Carole André, Paloma Moreno-Elgard (2018). Les nouvelles techniques de séquençage. Savoir & Comprendre. 3-4. 6- Illumina® (2020). Key differences between next-generation sequencing and Sanger sequencing, Comparison of Sanger Sequencing and NGS, Illumina (en ligne). https://www.illumina.com/science/technology/next-generation-sequencing/ngs-vs-sanger-sequencing.html?langsel=/us/ (consulté le 21/03/2020) 7- Martina Kujanová (2020). Higher-quality sequence data at the beginning of electropherogram - Modified sequencing primers, SEQme (en ligne) https://www.seqme.eu/en/magazine/Modified-sequencing-primers (consulté le 21/03/2020) | ||

| MR | Séquençage Illumina | |
|---|---|---|
Séquençage Illumina I. Objectifs Le séquençage Illumina est une méthode qui permet le séquençage de plusieurs centaines de millions de fragments d'ADN. L’application de cette « Next generation sequencing » (NGS) permet d’étudier la diversité microbienne dans l’environnement, de séquencer des génomes entiers, ou d’analyser les interactions entre ADN et protéines. Les domaines d’applications sont les suivants : la recherche sur le cancer, l’agrigénomique, les analyses criminalistiques, la FIV (Fécondation In Vitro), le DPI (Diagnostic Préimplantatoire), la découverte de virus ou la caractérisation de micro-organismes non cultivables 3. II. Principe Cette technologie se base d’abord sur l’acquisition d’une librairie1. Les brins d’ADN sont fragmentés en segments de 100 à 500 pb et sont couplés à leurs extrémités à des séquences particulières (une séquence permettant la liaison, des séquences index et une région complémentaire aux oligonucléotides de la plaque). Ces fragments sont amplifiés en PCR « bridge » en phase solide. Le séquençage est réalisé grâce à un émetteur fluorescent fixé aux nucléotides (un type de nucléotide est associé à un type de longueur d’onde qui lui est propre)3. La technologie Illumina s’appuie sur différentes techniques qui dont le SBS (sequencing by synthesis), la patterned flow cell technology et l’acquisition de données de plusieurs téraoctets.
Figure 1: Brins d'ADN fragmentés III. Mode opératoire Les fragments d’ADN sont déposés sur une plaque recouverte de deux types d’oligonucléotides chacun identiques à l’une des extrémités des brins d’ADN. L’ADN complémentaire à ces fragments est synthétisé en se servant des oligonucléotides de la plaque comme support initiateur (Figure 2). Les fragments primaires d’ADN sont dénaturés puis lavés grâce à un tampon spécifique2. Le brin se plie et s’hybride au second type d’oligonucléotides présent sur la plaque 3. Il y a synthèse du brin antisens formant ainsi un pont double brin. Le pont est dénaturé donnant ainsi un brin linéaire sens et un brin linéaire antisens identique au brin d’origine (Figure 3). Cette étape est répétée de nombreuses fois. Après l’amplification, les ponts sont coupés et la partie reverse est lavée grâce à un tampon2. Pour assurer la synthèse du brin dans le bon sens, les trois premiers nucléotides attachés aux oligonucléotides du tapis sont « bloqués ». L’ajout de l'amorce de séquençage permet de guider la polymérisation réalisée avec des nucléotides marqués avec une sonde fluorescente (Figure 4). Les clusters sont excités à une longueur d’onde donnée entrainant ainsi l’émission d’un signal lumineux (ce type de séquençage est appelé « séquençage par synthèse »). Le nombre de cycle détermine la longueur du fragment. La longueur d’onde et l’intensité du signal émis déterminent le nucléotide associé. Tous les brins identiques sont lus en même temps. Une fois que la première lecture est terminée le brin dernièrement synthétisé est éliminé. L’index 1 est introduit et hybridé au brin puis « complété » par synthèse jusqu’à atteindre les oligonucléotides. Une fois complété, l’index 1 est lavé et les trois derniers nucléotides sont déprotégés1 (Figure 5). Le brin se plie alors et se lie au deuxième type d'oligo. L'index 2 est synthétisé de la même manière pour permettre la fixation de l'ADN polymérase qui élongue l'oligonucléotide formant ainsi un pont double-brin qui sera dénaturé, engendrant deux brins linéaires (Figure 6). Les extrémités 3’ sont bloquées et le brin sens est éliminé laissant seulement le brin antisens. La deuxième lecture est initiée par la fixation de l'amorce de séquence 2 permettant ainsi la synthèse du brin d'intérêt avec les nucléotides excitables. Ce brin nouvellement synthétisé est ensuite éliminé à son tour. IV. Présentation des résultats La synthèse des brins complémentaires aux brins fixés sur les oligonucléotides de la plaque entraîne pour chaque nucléotide qui se fixe, l’émission d’une longueur d’onde qui lui est particulière. Les fragments dont la synthèse a été enregistrée sont ensuite triés selon la séquence index qui leur a été attribué lors de la préparation des échantillons (séquence qui a elle aussi été synthétisée, une séquence est propre à un fragment d’ADN). Pour chaque échantillon, les séquences possédant des schémas de nucléotides similaires sont regroupées. Les brins sens et anti sens sont appariés permettant ainsi la construction de séquences continues. Ces séquences continues sont ensuite alignées par rapport au génome de référence 5. Tout ceci est réalisé grâce à un logiciel interne à l'entreprise : BaseSpace 6. V. Interprétation des résultats L’interprétation des résultats dépend de l’objectif du chercheur. Selon l'absence ou la présence de la séquence identifiée on peut déterminer l'implication d'un gène dans une maladie. Grâce aux transcrits obtenus (dans le cas d'une rétro-transcription d'ARN), on peut aussi identifier des gènes et, par la même occasion, estimer l'abondance des transcrits afin de quantifier la transcription d'un gène. Les séquences obtenues peuvent ensuite être utilisées pour réaliser les différents tests bioinformatiques dont le chercheur a besoin. VI. Intérêts et limites L’avantage de cette méthode est une amélioration dans la vitesse du séquençage en comparaison à une méthode de séquençage « Sanger ». De plus, l’exactitude du séquençage est estimée à 99.9%. L’équipement peut être coûteux (Hiseq1000 : 560k$), mais en comparaison à d’autres méthodes comme la méthode Sanger, le prix de réalisation du séquençage n’est en lui-même pas cher (10k$) 3. De plus, contrairement une nouvelle fois à la méthode Sanger, il n’est pas nécessaire de réaliser un clonage bactérien pour assurer l’efficacité du séquençage. Cependant, les séquences obtenues sont plus courtes du fait de la fragmentation de l’ADN. De plus, un génome de référence doit être établi au préalable, ce qui demande un temps de travail supplémentaire. La quantité de données obtenue (de l’ordre du téraoctet) est non négligeable 4. Il faut prévoir une grande capacité de stockage et faire un choix dans ce que l’on garde ou pas. La transmission des résultats en sera impactée.
VII. Références bibliographiques (1) Meyer M. and Kircher M. (2010). Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and sequencing. Cold spring Harb Protoc. (2) Hodges E., Rooks M., Xuan Z., Bhattacharjee A., Gordon BG., Brizuela L., McCombie WR., and Hannon GJ. (2009). Nat protoc. 4(6): 960-974. (3) https://www.illumina.com/technology/next-generation-sequencing/sequencing-technology.html (4) https://www.illumina.com/technology/next-generation-sequencing.html (5) Illumina Sequencing by Synthesis. Disponible sur: (6) Site web de la compagnie Illumina. Disponible sur : https://www.illumina.com/
| ||

| CH | Séquençage Nanopores | |
|---|---|---|
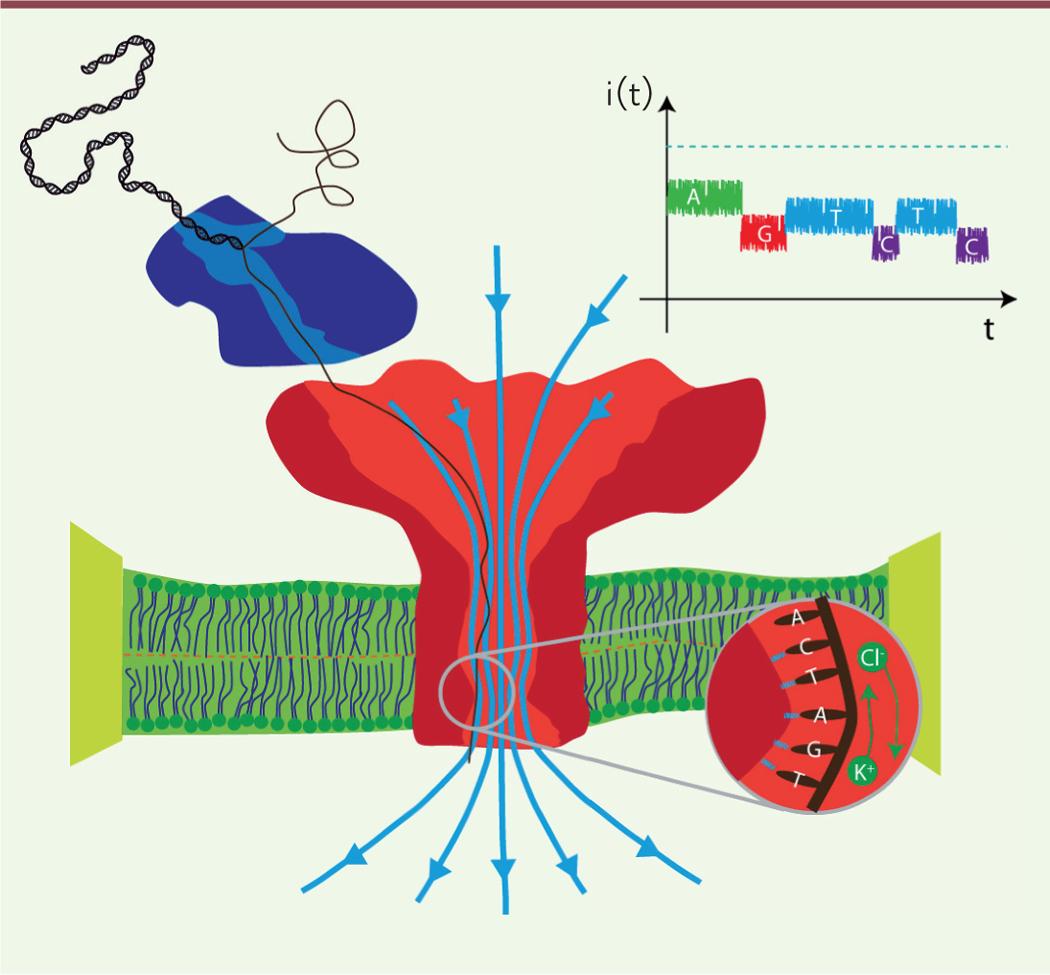
Séquençage Nanopores Objectifs Le séquençage Nanopore a été découvert en 19951 et a pour objectif de séquencer des échantillons d’ADN, c’est-à-dire de déterminer l’ordre dans lequel se trouvent les 4 bases Adénine (A), Cytosine (C), Thymine (T) et Guanine (G) des nucléotides sur le brin d’ADN. Principe Le principe du séquençage par Nanopore est de déterminer la séquence de la molécule d’ADN par l’utilisation de nanopores (pores avec un diamètre de l’ordre d’un nanomètre). Les nanopores peuvent être : - d’origine biologique en utilisant des protéines transmembranaires telles que les porines, - d’origine synthétique construits à partir de résidus de silicium2 ou de graphène3. Dans le cas du silicium, un trou de quelques nanomètres est partiellement comblé avec des couches d’ions déposées à l’aide de méthodes de sculpture par faisceau d'ions2. Le diamètre du pore est alors de l’ordre de 1 nm. Lorsqu’un nanopore est placé dans un milieu conducteur et soumis à un voltage, il est traversé par un courant électrique issu de la conduction d’ions. Ce courant est mesurable et dépend de : - la taille et la forme du nanopore, - la nature et concentration des particules traversant ou passant à proximité du nanopore. Le courant est ainsi modifié de manière caractéristique lorsque le pore est traversé ou obstrué par un nucléotide contenant les bases A, C, T ou G. En faisant passer un brin d’ADN par le nanopore, il est donc possible de déterminer la séquence de la molécule4. L’ADN étant double brin, il ne peut passer dans le nanopore que si l’hélice est ouverte. Pour cela, des enzymes comme l’hélicase ou l’ADN polymérase génétiquement modifiée sont utilisées pour se complexer avec le pore et ouvrir l’hélice d’ADN directement en direction du pore (Figure 1)5.
Figure 1 : Principe du séquençage par Nanopore biologique 5 L’hélice d’ADN est ouverte par une enzyme (en bleu), et le brin simple d’ADN peut ainsi passer à travers du nanopore (en rouge) qui se trouve au sein d’une membrane (en vert). Le courant ionique (flèches bleues) favorise le passage de l’ADN à travers le pore, entraînant ainsi un changement de vitesse de passage des ions, selon le nucléotide qui se trouve dans le pore (encerclé en bas à droite). Le graphique en haut représente l’identification des nucléotides, par rapport au courant mesuré, le pointillé bleu représentant le courant à vide. Mode opératoire Le séquençage par nanopore est une technique commercialisée par Oxford Nanopore Technologies, une entreprise qui a depuis lancé son premier dispositif portable de séquençage par nanopores. Afin de pouvoir effectuer ce séquençage, des kits de réactifs sont commercialisés avec la machine. Pour charger le séquenceur, il faut premièrement amorcer la cellule d’écoulement. Pour cela, 800 µl de tampon d'amorçage sont introduits dans la cellule via le pore d'amorçage, en évitant l'introduction de bulles d'air. Après avoir attendu 5 min, le couvercle du port d'échantillonnage SpotON est soulevé et le reste du tampon d’amorçage, soit 200 µl, est introduit dans la cellule d’écoulement via le pore d’amorçage6. L’échantillon d’ADN à séquencer est mélangé doucement par pipetage avant le chargement. Puis, 75 μl d’échantillon sont introduits goutte à goutte à la cellule d’écoulement via le pore d'échantillonnage SpotON préalablement ouvert6. Le séquençage est alors réalisé, les résultats pouvant être lu en direct sur l’ordinateur. Le principe de fonctionnement du séquenceur nanopore est résumé dans la Figure 2.
Figure 2 : Principe de fonctionnement du séquenceur MinION de l’entreprise Oxford Nanopore Technologies7
Présentation des résultats Le séquenceur donne le résultat sous forme d’une variation de courant déjà analysée. La séquence peut ainsi être directement lue (Figure 3). Figure 3 : Exemple de résultat de séquençage par nanopore8 À chaque nucléotide est associé un changement d’intensité caractéristique traduisant l’encombrement du canal lors de son passage (Figure 2). Pour déterminer de quelle base il s’agit, il suffit de comparer le niveau d’intensité électrique lors de son passage dans le canal à l’intensité électrique mesurée à vide. Ainsi : - la base A correspond à un courant d'environ 2 picoampères (pA). - la base G correspond à un courant d'environ 4 pA. - la base C correspond à un courant d'environ 6 pA. - la base T correspond à un courant d'environ 7 pA.
Interprétation des résultats L’interprétation des résultats dépend de ce que le chercheur veut faire. Il est possible de comparer ces résultats à des logiciels de bio-informatique, et ainsi d’identifier des patterns relatifs à certains gènes ou sites de fixation. Il est également possible de mettre en évidence des mutations entraînant par exemple une maladie génétique. Intérêts et limites Le séquençage nanopores a de nombreux avantages : - Il ne nécessite pas d’amplification préalable de l’ADN et permet donc d'éliminer toute erreur introduite au cours du processus d'amplification et de simplifier les manipulations. - Il permet le séquençage d’échantillons atteignant des longueurs allant jusqu’à 1Mb. Ainsi, il est possible de séquencer un génome complet grâce à cette méthode. - Il fait partie des méthodes de séquençage les plus rapides (entre 1 min et 72 h). Ce paramètre dépend de la taille et de la nature de la molécule à séquencer, mais aussi du type d’appareil utilisé9. - Les séquenceurs ONT sont moins chers, portables et de taille nettement plus réduite, par rapport aux plateformes de séquençage de deuxième génération, ce qui permet par exemple un séquençage directement sur le terrain10. Une des principales limites de cette technique est un taux d’erreur relativement élevé (2-15 %) par rapport à celui observé avec les technologies à lecture courte9. Afin de lutter contre ceci, les nanopores sont constamment améliorés pour favoriser, par exemple, un passage plus lent des nucléotides à travers le pore. De plus, les pores synthétiques sont de plus en plus utilisés, et de nouvelles méthodes sont élaborées afin de compenser cette faiblesse. Le séquençage nanopore par fluorescence5, par exemple, qui couple les nucléotides à des fluorescents, augmentant ainsi la précision d’identification des nucléotides, est en plein développement. Une autre limite de la méthode est la nécessité de disposer de quantités relativement importantes de matériel génétique9.
Références bibliographiques [1] Kasianowicz JJ, Brandin E, Branton D et Deamer DW. (1996). Characterization of individual polynucleotide molecules using a membrane channel. Proceedings of the National Academy of Sciences., 93 (24), p. 13770–3. [2] Cai Q, Ledden B, Krueger E, Golovchenko JA, et Li J. (2006). Nanopore sculpting with noble gas ions. J Appl Phys. 100(2), p. 24914-249146. [3] Garaj S, Hubbard W, Reina A, Kong J, Branton D, et Golovchenko J. A. (2010). Graphene as a subnanometre trans-electrode membrane. Nature, 467(7312), p. 190–193. [4] Raza S & Ameen A. (2017). Nano pore Sequencing Technology: A Review. International Journal of Advances in Scientific Research. 3 (8), 90. [5] Montel F. (2018) Séquençage de l’ADN par nanopores - Résultats et perspectives. Med Sci. 34 (2). 161-165 [6] Oxford Nanopore Technology. Priming and loading your flow cell. Disponible à : https://nanoporetech.com/#gns[searchValue]=Priming%20and%20loading%20your%20flow%20cell (consulté le 16/03/22) [7] Wang Y, Zhao Y, Bollas A, Wang Y et Fai Au K (2021) Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol. 39, 1348–1365 [8] Fuller C, Kumar S, Porel M, Chien M, Bibillo A, Stranges P, Dorwart M, Tao C, Li Z, Guo W, Shi S, Korenblum D, Trans A, Aguirre A, Liu E, Harada E, Pollard J, Bhat A, Cech C, et Ju J. (2016). Real-time single-molecule electronic DNA sequencing by synthesis using polymer-tagged nucleotides on a nanopore array. Proceedings of the National Academy of Sciences. 113 (19), 201601782. [9] Taishan H, Nilesh C, Dimitri M, et Anh D. (2021) Next-generation sequencing technologies: An overview. Human Immunology. 82(11), 801-811 [10] Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L, Bore JA, Koundouno R, Dudas G, Mikhail A, Ouédraogo N, Afrough B, Bah A, Baum JH, Becker-Ziaja B, Boettcher JP, Cabeza-Cabrerizo M, Camino-Sanchez A, Carter LL, Doerrbecker J, Enkirch T, Dorival IGG, Hetzelt N, Hinzmann J, Holm T, Kafetzopoulou LE, Koropogui M, Kosgey A, Kuisma E, Logue CH, Mazzarelli A, Meisel S, Mertens M, Michel J, Ngabo D, Nitzsche K, Pallash E, Patrono LV, Portmann J, Repits JG, Rickett NY, Sachse A, Singethan K, Vitoriano I, Yemanaberhan RL, Zekeng EG, Trina R, Bello A, Sall AA, Faye O, Faye O, Magassouba N, Williams CV, Amburgey V, Winona L, Davis E, Gerlach J, Washington F, Monteil V, Jourdain M, Bererd M, Camara A, Somlare H, Camara A, Gerard M, Bado G, Baillet B, Delaune D, Nebie KY, Diarra A, Savane Y, Pallawo RB, Gutierrez GJ, Milhano N, Roger I, Williams CJ, Yattara F, Lewandowski K, Taylor J, Rachwal P, Turner D, Pollakis G, Hiscox JA, Matthews DA, O'Shea MK, Johnston AM, Wilson D, Hutley E, Smit E, Di Caro A, Woelfel R, Stoecker K, Fleischmann E, Gabriel M, Weller SA, Koivogui L, Diallo B, Keita S, Rambaut A, Formenty P, Gunther S et Carroll MW.(2016) Real-time, portable genome sequencing for Ebola surveillance. Nature. 530, 228–232 | ||


| MP | SHERLOCK et DETECTR: 2 outils CRISPR | |
|---|---|---|
SHERLOCK et DETECTR : 2 outils CRISPR
I) Objectifs SHERLOCK (Specific High Sensitivity Reporter unLOCKing) et DETECTR (DNA Endonuclease Targeted CRIPR Trans Reporter) sont de nouvelles applications de la technologie CRISPR-Cas. Ce sont des outils de diagnostic basés sur une migration sur papier identifiant certaines signatures génétiques ou molécules cibles associées (Leitch, 2018) II) Principe Ces méthodes sont utilisées pour un diagnostic rapide. Cependant il faut avoir une connaissance de la séquence ciblée comme pour un CRISPR, il faut par exemple vérifier qu’elle est unique dans l’échantillon, afin de bien synthétiser l’ARN guide s’hybridant avec la cible. SHERLOCK permet, par exemple, d’identifier la présence ou non d’ADN tumoral dans les cellules sanguines de patients atteints de cancer (Zusi, 2018), de quantifier le stade de contamination dans le cas d’un virus (Zusi, 2018) ou de vérifier la présence d’un gène bactérien codant pour une résistance aux antibiotiques, un génome viral ou une mutation causant un cancer (Leitch, 2018) DETECTR possède le même principe de fonctionnement et a permis l’identification de différents types de Papilloma Virus (HPV), en détectant leur ADN dans les cellules humaines (Potenza, 2018).
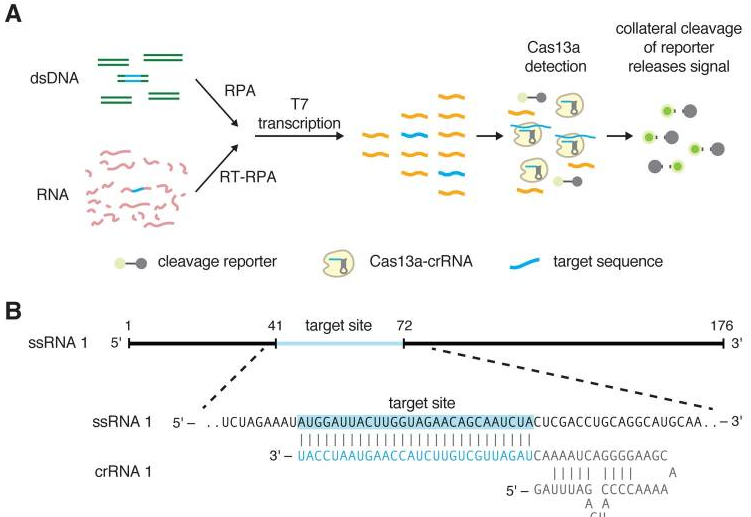
III) Mode opératoire Une étape préliminaire d’amplification isotherme par une polymérase recombinase (RPA) optimise le résultat de ces outils, en abaissant significativement la concentration minimale nécessaire à la détection de la cible. Cette étape va permettre l’hybridation des amorces avec la séquence cible, son élongation et son amplification à une température de 37-42 °C (Gootenberg et al, 2018) La technique SHERLOCK utilise la technique CRISPR avec la protéine Cas13 qui coupe de l’ARN ou de l’ADN à un endroit prédéfini. Pour cela, un ARN guide d’une trentaine de bases est synthétisé. La protéine Cas 13 cherche la séquence complémentaire de cet ARN ou ADN dans l’échantillon et, lorsqu’elle la trouve, la coupe. Un reporter (ARN synthétique) est ajouté dans le mix et est coupé (activité RNAse) si Cas13 trouve la séquence cible. Une molécule signal est alors émise (par exemple fluorescente) pour confirmer sa présence (Figure 1) (Zusi, 2018).
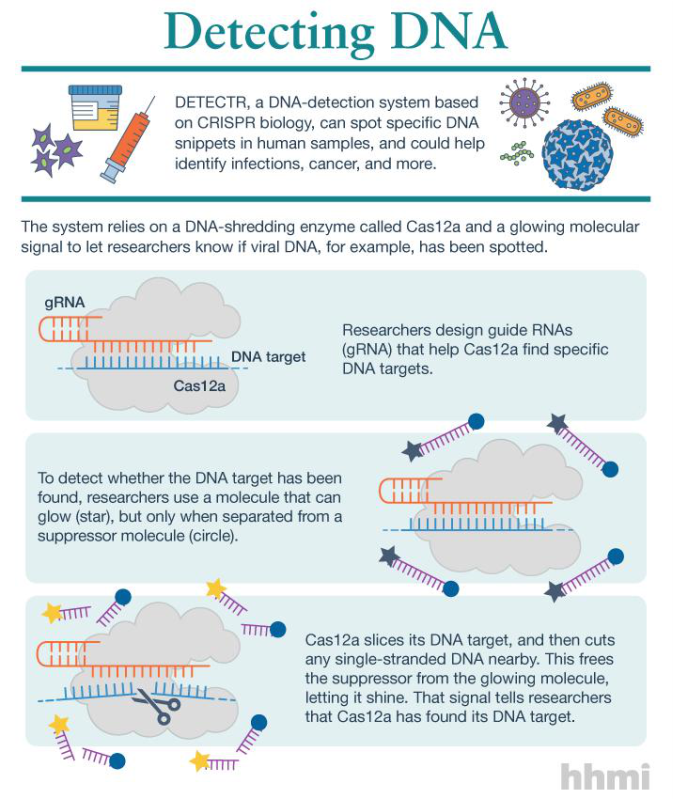
Figure 1 : Schéma de fonctionnement de cas13a et de l’émission du signal rendant compte du clivage de la séquence d’intérêt (Gootenberg et al, 2017) Le principe de fonctionnement de DETECTR (Figure 2) est le même que pour SHERLOCK, mais la protéine utilisée est l’enzyme Cas12a, et la cible est de l’ADN. Lorsque cette protéine détecte sa séquence cible et la coupe, elle présente une forte activité DNAse. Cette dernière est non-spécifique et découpe les monobrins d’ADN présents dans l’échantillon. Ainsi, des molécules d’ADN synthétiques monobrins sont utilisées comme marqueurs fluorescents. Elles sont constituées d’éléments suppresseurs liés aux marqueurs et introduites dans l’échantillon. Lorsque Cas12a a reconnu sa cible, ces monobrins sont découpés et il y a émission de fluorescence (Leitch, 2018).
Figure
2 : Schéma de fonctionnement de la méthode DETECTR à l’échelle moléculaire
(Zusi, 2018) IV) Présentation des résultats
Figure 3 : Photo présentant les bandelettes de migration de la méthode SHERLOCK. A gauche, se trouvent les papiers sur lesquels il n’y a pas eu de test effectué ; Au centre se trouvent les bandelettes avec une mesure négative. Enfin à droite des bandelettes affichant une mesure positive de la méthode SHERLOCK. (Zusi, 2018)
Figure 4 :
Identification des HPV type 16 et 18 dans des échantillons humains par PCR (à
droite) par DETECTR (à gauche) (Chen et al, 2018) V) Interprétation des résultats La présence d’une bande permet de savoir que la séquence cible a été reconnue par l’enzyme utilisée. VI) Intérêts et limites Ces outils de diagnostic sont faciles d’utilisation du fait qu’il n’y ait ni besoin de purifier, ni de dénaturer la séquence cible. Elles sont assez sensibles du fait de l’étape préliminaire d’amplification et rapides (moins d’1h30 pour la détection couplée des virus Zika et de la Dengue en utilisant SHERLOCK) (Gootenberg et al, 2018). Aujourd’hui, l’optimisation de l’utilisation des différentes enzymes Cas est en cours. En effet, l’étude des préférences de clivages de chaque enzyme Cas permettra la détection de plusieurs cibles à la fois dans le même échantillon. SHERLOCK deviendrait alors multiplexe. De plus, SHERLOCK nécessite assez peu de matériel. Cas 13 peut être lyophilisé, et est donc facilement transportable. Il suffit de mélanger SHERLOCK à l’échantillon pour détecter la séquence cible (Gootenberg et al, 2018). Cela peut être pratique pour des détections de virus rapides dans des populations à risques comme en Afrique vis-à-vis de Zika. Enfin, l’interprétation des résultats est très simple avec une lecture par présence ou non de bande. Cependant, ces outils restent performants pour des cibles dont on connait le séquençage complet. (Chen et al., 2018 ; Gootenberg et al., 2017) VII) Références bibliographiques CHEN, Janice S., MA, Enbo, HARRINGTON, Lucas B., DA COSTA, Maria, TIAN, Xinran, PALEFSKY, Joel M. et DOUDNA, Jennifer A. (2018) CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. In : Science. 15 février 2018. p. eaar6245. DOI 10.1126/science.aar6245. GOOTENBERG, Jonathan S., ABUDAYYEH, Omar O., KELLNER, Max J., JOUNG, Julia, COLLINS, James J. et ZHANG, Feng (2018) Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. In : Science. 15 février 2018. p. eaaq0179. DOI 10.1126/science.aaq0179. GOOTENBERG, Jonathan S., ABUDAYYEH, Omar O., LEE, Jeong Wook, ESSLETZBICHLER, Patrick, DY, Aaron J., JOUNG, Julia, VERDINE, Vanessa, DONGHIA, Nina, DARINGER, Nichole M., FREIJE, Catherine A., MYHRVOLD, Cameron, BHATTACHARYYA, Roby P., LIVNY, Jonathan, REGEV, Aviv, KOONIN, Eugene V., HUNG, Deborah T., SABETI, Pardis C., COLLINS, James J. et ZHANG, Feng (2017) Nucleic acid detection with CRISPR-Cas13a/C2c2. In : Science. 28 avril 2017. Vol. 356, n° 6336, p. 438‑442. DOI 10.1126/science.aam9321. LEITCH Carmen (2018), New Tools in the CRISPR Arsenal, S.1 LabRoots https://www.labroots.com/trending/cell-and-molecular-biology/8071/tools-crispr-arsenal consulté le 31/03/2018 POTENZA Alessandra (2018), New CRISPR tools can detect infections like HPV, dengue, and Zika, The Verge https://www.theverge.com/2018/2/15/17012866/crispr-detectr-sherlock-zika-dengue-hpv-diagnostic-tools consulté le 31/03/2018 ZUSI Karen (2018), SHERLOCK team advances its CRISPR-based diagnostic tool, S.1 BROAD Institute https://www.broadinstitute.org/news/sherlock-team-advances-its-crispr-based-diagnostic-tool consulté le 31/03/2018 | ||


| VN | Southern et Northern blots | ||
|---|---|---|---|
Technique d'analyse moléculaire par hybridation d'une sonde d'ADN connu marquée. | |||
| SC | Système CRISPR-dCas9 | ||
|---|---|---|---|
Systeme CRISPR-dCas9
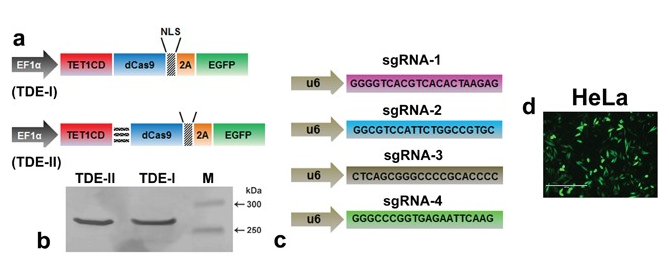
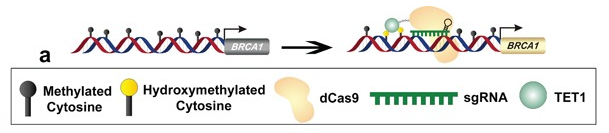
I. Objectifs Le système « CRISPR-defficient Cas9 » aussi appelé CRISPR-dCas9 (clustered regularly interspaced short palindromic repeats) est une méthode dérivée de la technique d’édition génomique CRISPR-Cas9, où l’activité catalytique de Cas9 a été désactivée. Elle est souvent utilisée pour la visualisation de génomes, des manipulations épigénétiques, l’analyse de composition de chromatine, et trouve donc de nombreuses applications en recherche médicale (Anton et al., 2018). Principe Les procaryotes possèdent un système de défense immunitaire adaptative basé sur des CRISPR-RNAs associés à des gènes Cas. Le système CRISPR/Cas a la fonction d’identifier une séquence cible et de guider les nucléases vers des acides nucléiques à lyser. Dans le cas de CRISPR-dCas9, le site catalytique de Cas9 est muté simultanément sur deux résidus spécifiques, ce qui entraîne l’inhibition de l’activité endonucléase de l’enzyme (Anton et al., 2018). Pour illustrer une application du système CRISPR-dCas9, nous analyserons le cas du ciblage de translocation Ten-Eleven I (TET-I) par CRISPR-dCas9 pour la déméthylation sélective de l'ADN au niveau du promoteur de BRCA1. En effet, L’ADN sur-méthylé sur les gènes suppresseurs de tumeurs en lien avec l'extinction de la transcription est une caractéristique commune des cancers. Il existe des enzymes de déméthylation de la famille des dioxygénases des TET. Afin d’étudier le rôle des enzymes TET qui effacent les marques de méthylation, le domaine catalytique de TET-I (TET-I-CD) est fusionné à dCas9. Cette protéine combinée à des sgRNA (single RNA) permet de cibler le promoteur de BRCA1 (Breast Cancer 1), un gène suppresseur de tumeur. Ce gène présente une hyperméthylation sur son promoteur qui provoque l’extinction du gène dans le cancer du sein et de l’ovaire (Choudhury et al., 2019). Mode opératoire La fusion de la protéine avec la dCas9 se fait en assemblant séquentiellement les séquences codantes des protéines souhaitées grâce à des enzymes de restrictions et de ligations. Dans notre cas, les séquences de TET-I catalytic domain (TET-I-CD) associées à l’extrémité N-terminale de la dCas9 sont introduites dans un plasmide. La protéine dCas9 est marquée par le rapporteur fluorescent EGFP (Enhanced Green Fluorescent Protein). Deux protéines de fusion ont été synthétisées, TDE-I et TDE-II (TDEs), avec respectivement une absence ou une présence de séquence de liaison entre TET-I-CD et dCas9 (Figure 1a). L’expression des protéines TDEs dans des cellules HeLa est vérifiée par Western blot (Figure 1b). La protéine de fusion fait au total 269 kDa. Le plasmide est transfecté avec des sgRNA spécifiques à des séquences différentes (Figure 1c). Ils guident la dCas9 sur 4 loci du promoteur BRCA1, mettant en contact la protéine TET-I avec des cytosines méthylées différentes. Les positions favorisent la déméthylation d’un plus ou moins grand nombre de cytosines. Enfin, une transfection du plasmide est réalisée pour contrôler son introduction dans les noyaux, ici, de cellules HeLa (Figure 1d) (Choudhury et al., 2019).
Figure 1 : Conception du système CRISPR-dCas9 pour le ciblage du promoteur BRCA1 par TET-I (Choudhury et al., 2019). Présentation des résultats Le complexe TET-I-dCas9 va cibler des régions spécifiques du promoteur de BRCA1 grâce aux sgRNA sélectionnés (Figure 1c). L’enzyme TET-I-CD va alors se fixer aux cytosines méthylées et effectuer une déméthylation sélective de ces sites du promoteur de BRCA1 (Figure 2). La mise en évidence de la déméthylation des cytosines par le complexe se fera par mesure de l’activité transcriptionnelle du gène BRCA1, par PCR quantitative (qPCR).
Figure 2 : Représentation de la régulation positive de l’expression du gène BRCA1 par hydroxyméthylation spécifique des loci par le complexe TET-I-dCas9. (Choudhury et al., 2019).
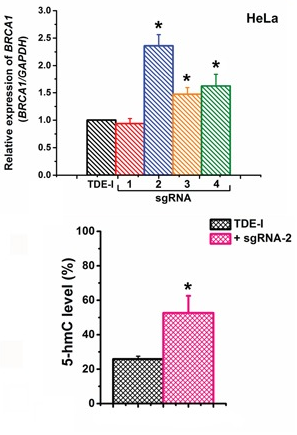
Interprétation des résultats Le complexe TET-I-dCas9 déméthyle la cytosine lorsque celui-ci est transfecté avec les sgRNA 2, 3, 4, comme le montre l’expression du gène BRCA1 par rapport à l’expression du témoin. Le système dCas9-TET1 va se placer sur le promoteur, il réalise son rôle de transporteur avec précision (Figure 3a). La déméthylation qui se traduit par une hydroxylation de la 5-mC prouve la précision du système avec l’ARN guide 2 (Figure 3b).
Figure 3 : Effets du ciblage du promoteur BRCA1 par TET-I sur l’expression génétique et l’enrichissement en 5-hydroxyméthylation (5-hmC). (Choudhury et al., 2019) a - L’expression du gène BRCA1 après traitement des cellules HeLa avec TDE-I et différents sgRNA a été déterminée par qPCR. b - Le promoteur BRCA1 a été enrichi en 5-hmC après transfection des cellules HeLa avec TDE-I et le sgRNA-2.
Intérêts et limites Cette méthode a l’avantage d’être peu coûteuse et facile à mettre en place. De plus, elle possède une grande efficacité et une certaine spécificité envers ses cibles. C’est un outil de grand intérêt dans la compréhension des dynamiques autour de l’architecture nucléaire lors des différenciations cellulaires et des cycles cellulaires. L’édition épigénomique basée sur CRISPR-dCas9 permettra à long terme d’identifier les voies de régulation de maladies causant des changements à partir d’effets indirects en pathologie humaine. (Anton et al., 2018) Références bibliographiques Anton T, Karg E, Bultmann S. (2018) Applications of the CRISPR/Cas system beyond gene editing. Biology Methods and Protocols. 1–10. https://doi.org/10.1093/biomethods/bpy002
Choudhury SR, Cui Y, Lubecka K, Stefanska B, Irudayaraj J. (2019) CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget. 7(29):46545-46556. https://www.oncotarget.com/article/10234/text/ | |||
| TL | Système de Recombinaison Cre-LoxP | ||
|---|---|---|---|
Système de Recombinaison Cre-LoxP Objectifs Avant le développement de la technique de Recombinaison Cre-LoxP, la production de modèles vivants avec des gènes inactivés (Gene KnockOut) n’était pas possible si les gènes concernés étaient impliqués dans le développement embryonnaire. Il était également impossible de réaliser un KO dans un tissu ou un groupe de tissus particuliers. Le Système de Recombinaison Cre-LoxP (Nagy, 2000) est une technique qui permet une délétion, inversion ou translocation contrôlée dans l’espace et le temps. Elle est donc employée pour générer des lignées d’animaux présentant des KO tissu-spécifiques ou inductibles chimiquement en temps voulu et en observer les effets. Elle est notamment utilisée pour étudier la différenciation cellulaire (Dor et al, 2004) ou le rôle de protéines identifiées comme facteurs de risque pour des pathologies (Fujioka et al, 2011).
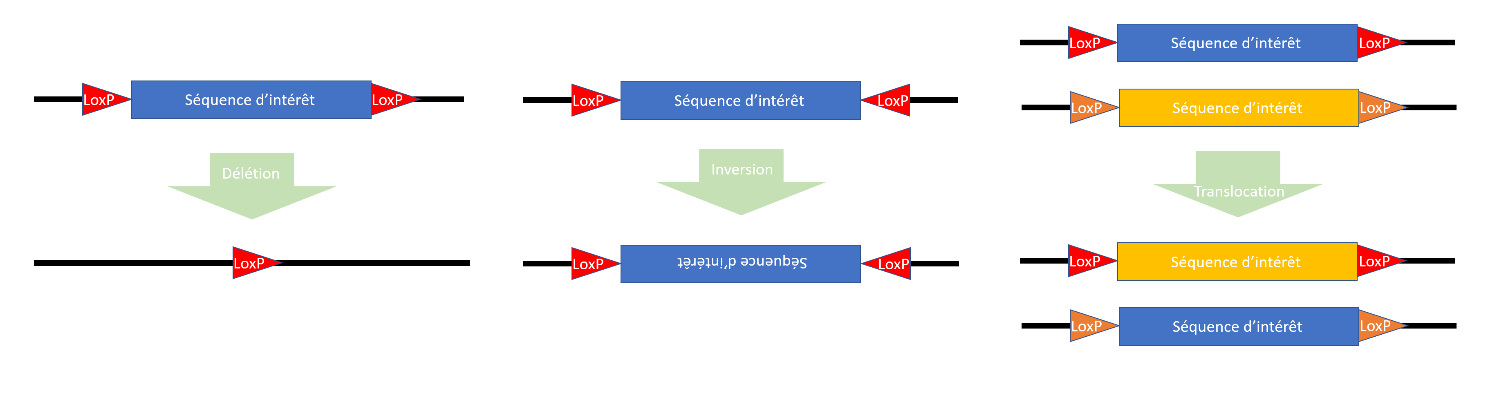
Principe La Recombinaison Cre-LoxP tire son nom de l’enzyme Cre Recombinase dérivée du Bactériophage P1 et qui a pour fonction d’exciser un fragment d’ADN situé entre 2 séquences LoxP. Grâce à l’asymétrie de la séquence LoxP, l’enzyme peut soit catalyser la délétion, l’inversion ou la translocation de la séquence ciblée :
Figure 1 : Action de la Cre Recombinase selon la position des séquences LoxP Le « floxing » (de l’anglais flanked by LoxP) se réalise avec CRISPR-Cas9 auquel la séquence LoxP. Une fois les séquences d’intérêt floxées, la Cre Recombinase dans les cellules de 3 façons : -l’introduire dans les cellules à l’aide d’un virus ; -insérer le gène de la Cre Recombinase sous contrôle du promoteur d’un gène dont l’expression dans la cellule cible ; -insérer un gène de Cre Recombinase ligand-dépendante (souvent un récepteur œstrogène sensible au tamoxifène) sous contrôle d’un promoteur non-spécifique (house-keeping gene). Mode opératoire
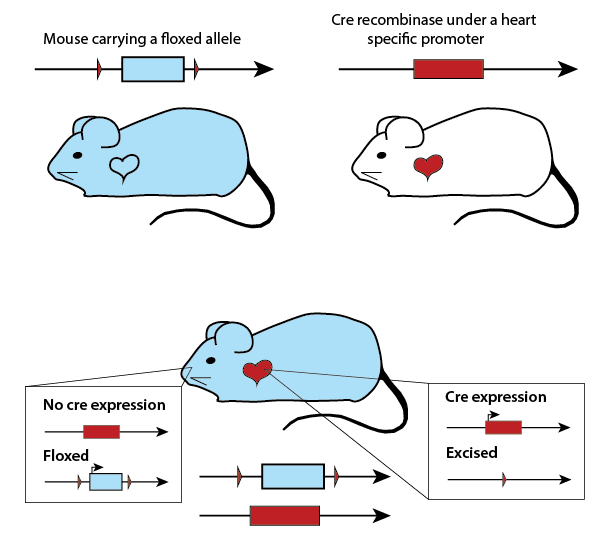
Les lignées générées sont de type Flox-Flox XXX Cre (ou FFXXXCre) où XXX est le gène dont le promoteur est utilisé pour faire exprimer la Cre Recombinase. La Cre Recombinase sera donc exprimée dans les cellules ciblées et délètera, inversera ou transloquera le gène floxé. Il est également possible d’induire l’expression de Cre Recombinase par un stimulus chimique induisant la transcription du gène. Présentation des résultats
Figure 2 : Délétion tissu-spécifique d’un allèle floxé par co-expression de Cre-Recombinase (MGI, Jax) Interprétation des résultats Le succès de l’opération peut être quantifié par PCR : -pour vérifier une délétion, l’amorce utilisée est complémentaire de la séquence que constituent les deux parties précédemment autour de la séquence d’intérêt et maintenant liées -pour une inversion ou translocation, utilise complémentaire de la séquence que forme la séquence d’intérêt avec la séquence qui entoure dans son nouveau locus. En cas de succès, l’hybridation est possible et la polymérisation a lieu. es échantillons sur Southern Blot déterminer le génotype des individus et le succès de la méthode. Intérêts et limites Cette méthode permet d’étudier l’influence de mutations inactivant des protéines et est du coup très utilisée dans l’étude de pathologies, notamment quand le facteur de risque identifié est exprimé seulement dans un certain type cellulaire. Cependant, cette méthode nécessite de connaître ou identifier un promoteur strictement tissu-spécifique. De plus, la création de lignées génétiquement modifiées avec des gènes supplémentaires est un processus coûteux en temps et argent. Références bibliographiques Nagy, A. (2000). Cre recombinase: the universal reagent for genome tailoring. Genesis. 26 (2) 99-109 Dor, Y., Brown, J., Martinez, O., Melton, DA. (2004) Adult pancreatic b-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 429 (6987) 41-46 Fujioka, M., Tokano, H., Fujioka, KS., Okano, H., Edge, ASB. (2011) Generating mouse models of degenerative diseases using Cre/lox-mediated in vivo mosaic cell ablation. The Journal of Clinical Investigation. 121 (6) 2462-2469 | |||