Wiki des méthodes de biologie moléculaire et cellulaire
Glossaire collaboratif des méthodes expérimentales de biologie moléculaire et cellulaire. Ce glossaire est réalisé par les étudiants du module "Méthodes expérimentales de biologie moléculaire et cellulaire" sous la supervision de l'équipe de Génétique Animale.
Spécial | A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z | Tout
C |
|---|

Chromatin Immunoprecipitation (ChIP) | ||
|---|---|---|
Chromatin Immunoprecipitation (ChIP)
1. Objectifs
L’immunoprécipitation de la chromatine est une méthode qui permet l’étude des protéines interagissant avec la molécule d’ADN. On obtient une représentation des interactions protéine–ADN qui ont lieu dans le noyau de la cellule vivante ou dans les tissus (in vivo). Elle est en général utilisée pour localiser des sites de fixation de facteurs de transcription, des sites associés avec des protéines de la chromatine, ou encore avec des histones possédant des modifications post-traductionnelles spécifiques.
2. Principe
Cette technologie se base d’abord sur la préparation de complexes protéine/ADN par action du formaldéhyde sur les cellules ou tissus puis sur l’immunoprécipitation de la chromatine en utilisant des anticorps spécifiques d’une protéine d’intérêt (présumée liée à l’ADN).
3. Mode opératoire
- La première étape est la formation de liaisons covalentes in vivo entre les protéines et l’ADN. En général, du formaldéhyde est ajouté sur des cellules, mais l’utilisation des UV ou de bleu de méthylène est aussi pratiquée. - Les cellules ainsi traitées sont ensuite lysées par sonification ou utilisation d’enzymes de restriction et des petits fragments d’ADN pontés (d’environ 200 à 500 paires de bases) sont obtenus. - On procède ensuite à l’immunoprécipitation de la chromatine pontée pour obtenir des complexes ADN/protéine purifiés grâce à un anticorps dirigé contre la protéine étudiée. Ceci permet de sélectionner les complexes du très grand nombre des autres fragments. Il est nécessaire d’avoir des anticorps d’une excellente qualité pour que la technique fonctionne correctement. - L’ADN précipité est ensuite purifié par chauffage pour annuler la réticulation par le formaldéhyde et l’ADN peut être séparé des protéines. Celles-ci sont digérées par la protéinase K. On obtient ainsi une collection de fragments d'ADN d'assez courte taille et dont on sait qu'ils interagissent avec une protéine d'intérêt (celle sélectionnée par l'anticorps).
Figure 1 : Mode opératoire de la méthode ChIP (Orlando, 1997) - On analyse ensuite l’ADN collecté grâce à une amplification par PCR et/ou à des puces à ADN en fonction de si la région du génome ciblé est connue.
4. Intérêts et limites
L’avantage majeur de la méthode ChIP est qu’elle s’effectue in vivo, c’est-à-dire que l’information tirée de cette expérience provient directement d’une analyse faite sur les cellules vivantes et l’immunoprécipitation permet de récupérer les protéines voulues ainsi que toutes les régions d’ADN auxquelles elles étaient liées lors du pontage initial au formaldéhyde. Cependant, malgré son succès la technique ChIP présente aussi des limites. Tout d’abord la résolution est limitée, on ne peut pas déduire la position précise du site de fixation de la protéine sur l’ADN, on identifie seulement une région de 200-300 pd dans laquelle se trouve le site de fixation ; on montre que la protéine se fixe en amont de tel gène mais sans que l’on sache où exactement. Une autre limite de la technique est que seules les protéines pour lesquelles on dispose d’anticorps peuvent être étudiées en ChIP. De plus les anticorps ont souvent le potentiel de réagir avec d'autres protéines nucléaires, malgré leur haute spécificité.
5. Présentation des résultats
L’étape clé de la technique de ChIP est la caractérisation de la région précise de l’ADN génomique qui fixe la protéine d’intérêt. On retrouve deux méthodes de caractérisation. La première est la technique par PCR en utilisant pour amorces des oligonucléotides correspondant aux gènes d’intérêt. Elle permet un enrichissement en séquences d’ADN reconnues par la protéine. Ainsi, la présence ou l’absence de séquence amplifiée révèle si oui ou non la protéine d’intérêt était liée à cette séquence d’ADN dans les cellules à partir desquelles la chromatine traitée au formaldéhyde a été isolée. Cependant, cette technique ne peut être utilisée que pour identifier des fragments connus et vérifier leur présence ou leur absence. On peut aussi caractériser les fragments par la technique Slot Blot.
Figure 2 : Présentation des résultats de la méthode ChIP (Kuo et Allis, 1999)
Quand on veut déterminer où la protéine est liée sur un génome à grande échelle, on peut utiliser une puce à ADN (Chip on chip). Dans cette approche, l’ADN total isolé de la cellule et l’ADN lié avec la protéine sont marqués chacun avec un fluorophore différent (dans la suite on parlera du fluorophore rouge pour l’ADN lié et du vert pour l’ADN total). Les deux ADN marqués sont mélangés et mis à hybrider sur la puce à ADN. C’est une approche très puissante puisqu’elle permet d’examiner simultanément tous les sites potentiels d’un génome entier sans qu’aucune connaissance préalable des sites potentiels ne soit nécessaire.
6. Interprétation des résultats
Dans le cas de l’analyse par PCR, si la protéine est fixée à la région de l’ADN attendue, alors l’ADN correspondant sera immunoprécipité lors de la ChIP et l’amplification sera possible. Deux contrôles importants sont nécessaires à ce stade. Le premier est d’inclure aussi deux amorces qui ciblent une autre région de l’ADN sur la laquelle la protéine étudiée est connue pour ne pas se fixer (aucune amplification ne devrait être observée ; c’est un contrôle négatif). D’autre part, on effectue une PCR sur un échantillon qui n'a pas subi une immunoprécipitation par des anticorps avec des amorces spécifiques et non spécifiques. Si les amorces PCR amplifient de la même façon leur cible, alors on devrait obtenir une amplification à peu près identique dans les deux cas. Ce contrôle permet de s’assurer que cette différence est significative d’une différence d’abondance des matrices et pas d’une différence d’efficacité entre les deux PCR. Après amplification, l’ADN est analysé par Slot Blot ou par Southern Blot. Dans le cas de l’analyse par puce à ADN, les zones de la puce à ADN qui ont un fort signal rouge sur vert correspondent aux séquences ayant fixé la protéine. Les zones de la puce à faible signal rouge sur vert sont les régions de l’ADN qui ne fixent pas la protéine étudiée.
7. Références bibliographiques
Carey, Michael F, Craig L Peterson, and Stephen T Smale. 2009. “Chromatin Immunoprecipitation (ChIP).” In Transcriptional Regulation in Eukaryotes: Concepts, Strategies, and Techniques, 2nd edition. Vol. 4. NY USA: CSHL Press. “Chip on Chip — Wikipédia.” 2015. July 3. http://fr.wikipedia.org/wiki/Chip_on_chip. Gilmour, David S, and John T Lis. 1985. “In Vivo Interactions of RNA Polymerase II with Genes of Drosophila Melanogaster,” Molecular and Cellular Biology, 5 (8): 2009–18. Gilmour, David S, and John T Lis. 1986. “RNA Polymerase II Interacts with the Promoter Region of the Noninduced hsp7O Gene in Drosophila Melanogaster Cells,” Molecular and Cellular Biology, 6 (11): 3984–89. “Immunoprécipitation — Wikipédia.” 2014. October 12. http://fr.wikipedia.org/wiki/Immunopr%C3%A9cipitation. Kuo, Min-Hao, and C. David Allis. 1999. “In Vivo Cross-Linking and Immunoprecipitation for Studying Dynamic Protein:DNA Associations in a Chromatin Environment,” November, Methods 19 edition, sec. 425-433. Locker, D. 2015. “‘ChIP on Chip’ Ou Comment Suivre La Technologie Des Gènes.” Accessed March 20. http://daniel.locker.perso.sfr.fr/article%20vulgarisation/CHIPonCHIP4.pdf Orlando, Valerio, Helen Strutt, and Renato Paro. 1997. “Analysis of Chromatin Structure by in Vivo Formaldehyde Cross-Linking,” METHODS: A Companion to Methods in Enzymology 11 edition. Watson, James, Tania Baker, Stephen Bell, Alexander Gann, Michael Levine, and Richard Losick. 2009a. “L’approche ChIP-Chip: La Meilleure Pour Identifier Des Enhancers.” In Biologie Moléculaire Du Gène, 6e edition. Pearson Education France. Watson, James, Tania Baker, Stephen Bell, Alexander Gann, Michael Levine, and Richard Losick. 2009b. “L’immuno-Précipitation E La Chromatine Permet de Détecter L’association Des Protéines Avec l’ADN Dans La Cellule.” In Biologie Moléculaire Du Gène, 6e edition. Pearson Education France.
| ||


CHROMATOGRAPHIE EN PHASE LIQUIDE ET SPECTROMETRIE DE MASSE (LC-MS) | |||
|---|---|---|---|
I) Objectifs LC-MS, tirée de l’anglais Liquid Chromatography – Mass Spectrometry, est une technique de laboratoire de chimie analytique pour l'identification, la quantification et l'analyse de masse de nombreuses substances. Comme son nom l’indique, elle combine les performances de la chromatographie en phase liquide et de la spectrométrie de masse. Ce couplage est aujourd’hui un des plus puissants utilisés par les chimistes et biochimistes en matière d’analyse qualitative (Arpino, 2009). La technique LC/MS peut être utilisée dans l'industrie pour détecter les contaminants à l'état de traces, mais aussi en industrie cosmétique et pharmaceutique, pour détecter des impuretés. Sur le plan environnemental, la LC-MS est utilisée pour l'analyse de divers échantillons tels que le sol, l'eau potable ou les eaux usées, l'air et les boues (détection de pesticides, d’herbicides). L’utilisation se fait aussi dans le domaine biomédical pour identifier des protéines, des lipides…. II) Principe Cette technique consiste en la succession de deux étapes :
III) Mode opératoire La LC-MS repose principalement sur l’utilisation d’un système de chromatographie liquide à haute performance (HPLC) et d’un système de spectrométrie de masse (MS). Le système HPLC permet la séparation des différents composants d’un échantillon liquide. En utilisant une phase stationnaire hydrophobe et une phase mobile polaire, il y a migration différentielle dans la colonne de chromatographie. Les molécules arrivent tour à tour à travers un PDA (photodiode array = réseau de photo diode) où le temps de rétention du composé dans la colonne ainsi que son absorbance vont pouvoir être mesurés (obtention d’un Dt, lmax). Les molécules migrent ensuite vers le système MS. Le système MS permet l’ionisation des molécules, par électrospray (ESI), ou par ionisation chimique à pression atmosphérique (APCI). Une fois rentrées dans le spectromètre de masse, les molécules chargées se dirigent ensuite dans un quadrupôle constitué de quatre tiges de charges, alternant rapidement entre charges positives et négatives. Cela induit des oscillations des ions. Seuls les ions qui n’heurtent pas les tiges de charges traversent le système et frappent le détecteur à l’extrémité du système. En fonction de la charge enregistrée par le quadrupôle à l’instant t de l’impact de la particule ionisée et des équations de mouvements, l’analyseur détermine le poids moléculaire (massenombre de charge=mz) du composé (Ying, 2022).
Figure 1 : Schéma du fonctionnement d’une LC-MS (Kailasam, 2022 d’après Cwszot, Dagui1929, Yassine Mrabet and Cas Ju reproduced under the Creative Commons CC0 1.0 Universal Public Domain Dedication license.)
Figure 2: Schéma d’un quadrupôle (Paul J. Gates, 2014, University of Bristol) IV) Présentation des résultats Une analyse LC-MS contient des informations sur le temps de rétention (RT) distinct pour chaque molécule, dans le chromatogramme, le rapport masse sur charge (m/z) dans le MS et l'abondance relative des ions pour chaque ion particulier. Les signaux MS détectés dans toute la gamme de séparation chromatographique sont formatés dans une carte tridimensionnelle, qui définit les données d'un seul passage LC-MS, comme le montre la Figure 3.
A. Figure 3 : Analyse LC-MS à la sortie du MS. Fig3.A (Tsai et al, 2016), Fig 3.B (Zhou et al, 2012) V) Interprétation des résultats L’aire sous le pic du spectre de rétention, permet de déterminer la concentration en analyte, par utilisation d’un pic de référence de l’isotope correspondant de concentration connue. A partir de la position du pic issu du spectre de masse, la masse de l’ion moléculaire est déterminée par comparaison aux tables. Lorsqu’un groupe de pics est observé sur le spectre de masse (m/z), cela correspond à un ensemble d’isotopes. VI) Intérêts et limites Avantages : LC-MS est une technique de haute sensibilité qui permet l’analyse de composés polaires, apolaires, d’ions et de molécules thermolabiles. Cette analyse simultanée induit ainsi une réduction du temps et du coût de la réalisation. De plus, selon ce que les chercheurs souhaitent montrer, par la diversité des colonnes à chromatographie, il est possible d’obtenir une séparation plus ou moins importantes des composés entre eux. Elle permet également de déterminer la masse moléculaire d’analytes de taille inférieure à 1 000 Da à des analytes de taille supérieure à 100 000 Da. Pour finir, en couplant les deux informations obtenues par la LC-MS (masse moléculaire et lmax), les chances d'identifier des mélanges complexes ainsi que des composés inconnus sont augmentées. Limites : L’appareillage de la LC-MS et l’analyse ultérieure des résultats peut être onéreux selon les revenus des laboratoires l’utilisant. Il y a une variabilité importante des spectres obtenus en sortie de LC, ce qui pourrait gêner l’identification. Les isomères ne peuvent pas être distingués. Leur masse est la même, mais pas leur structure. Le spectromètre de masse est un détecteur destructif, il faut donc faire attention lors de la manipulation d'échantillons qui peuvent ne pas être facilement disponibles. Enfin, l'analyse d'échantillons instables par LC-MS peut s'avérer difficile (Kailasam, 2022). VII) Références bibliographiques Arpino P (2009) Couplages chromatographiques avec la spectrométrie de masse. Techniques d’analyse. III (P1492). 1-24. doi: 10.51257/a-v1-p1492 Gates P (2014) Quadrupole Mass Analysis. UNIVERSITY OF BRISTOL. Consulté le 20 février 2023. http://www.chm.bris.ac.uk/ms/quadrupole.xhtml Kailasam S (2022) LC-MS – What Is LC-MS, LC-MS Analysis and LC-MS/MS. Technology Networks. Analysis & Separations. Consulté le 28 février 2023. https://www.technologynetworks.com/analysis/articles/lc-ms-what-is-lc-ms-lc-ms-analysis-and-lc-msms-348238#D2 Tsai T-H, Wang M, Ressom HW (2016) Preprocessing and Analysis of LC-MS-Based Proteomic Data. In K Jung, ed, Statistical Analysis in Proteomics. Springer New York, New York, NY, pp 63–76 Ying Z (2022) Qu’est-ce que la LC-MS. ANTITECK. Consulté le 20 février 2023. https://antiteck.com/fr/qu%27est-ce-que-lc-ms/ Zhou B, Xiao JF, Tuli L, Ressom HW (2012) LC-MS-based metabolomics. Mol BioSyst 8: 470–481 Réalisé par : DUFRENE Marjorie, GONAND Alix, POIRIER-COUTANSAIS Auriane, 2023
| |||


 B.
B.
Chromosome Conformation Capture | ||
|---|---|---|
Chromosome conformation capture (3C)
I) Objectifs La Capture de Conformation Chromosomique (3C) est une technique décrite pour la première fois par Job Dekker et ses collègues en 2002, qui permet l’étude de la structure 3D de la chromatine. Elle fournit des informations sur la structure 3D et les interactions des chromosomes in vivo à une échelle de quelques dizaines à quelques centaines de paires de kilobases (Vernet, 2013). Elle a pour but d’étudier l’organisation spatiale du génome, paramètre de contrôle de l’expression des gènes ainsi que sa dynamique au cours de différents processus biologiques tels que le cycle cellulaire, la mitose, la méiose…
II) Principe Cette méthode permet la détection des interactions chromosomiques, c’est-à-dire des contacts physiques directs ou indirects (médiés par des complexes ribonucléoprotéiques) entre une séquence cible et un ensemble de séquences prédites ou connues pour interagir avec la cible. Elle est applicable sur des populations cellulaires de nombreux organismes, allant des bactéries à l’Homme. III) Mode opératoire La méthode 3C nécessite deux étapes principales: la génération de produits de ligation et leur détection (Fig.1). Dans la première étape, les cellules sont fixées au formaldéhyde, les noyaux sont isolés, et les segments de chromatine spatialement proches sont réticulés de manière covalente au niveau d’un cross-link. Il peut s’agir de liaisons intra-chromosomiques ou inter-chromosomiques (Umlauf, 2015). Les fragments de chromatine à analyser sont ensuite coupés par des enzymes de restriction spécifiques, et subissent une ligation de manière à rabouter ensemble les portions de chromatine reliées par le cross-link. Puis, les produits de ligation sont purifiés. La deuxième étape consiste en une PCR (Hagège et al, 2007). Pour la méthode 3C, une vérification des interactions entre une séquence cible précise et des séquences prédites ou connues est réalisée. Grâce à cela, une PCR est conçue en choisissant les amorces spécifiques correspondantes (Gerosa, 2012). Deux types de PCR sont utilisés : la PCR semi-quantitative qui se base sur une électrophorèse sur gel, ou la PCR quantitative en ayant recours à des amorces fluorescentes.
Figure 1: Étapes de la méthode Capture de Conformation Chromosomique (Moindrot, 2012). IV) Présentation des résultats L’analyse de la PCR repose sur l’étude de l’intensité des bandes des amplicons qui reflète directement l’abondance du produit de ligation, et donc la proximité des deux segments de chromatine dans le noyau ou le cytoplasme.
Figure 2 : Révélation de la PCR (Dekker et al,
2002). Pour s’assurer de la pertinence des résultats, un contrôle est réalisé dans lequel est construit un chromosome bactérien contenant les séquences d’ADN des produits de ligation supposés (Fig.2). L’amplification par PCR permet d’analyser les cross-links (Hagège et al, 2007). Cela permet de s’assurer que les signaux obtenus ne sont pas dus à des problèmes liés à la technique : non amplification d’un fragment lors de la PCR dû à un mauvais choix d’amorce, par exemple, ou à une trop faible quantité d’ADN introduite. Sur ces graphiques, les interactions du fragment 6 (fragment cible) avec les fragments 5 et 13 sont étudiées. Seul le produit de ligation associant les fragments 5 et 6 est amplifié, alors que ce fragment est amplifié dans le contrôle. Même si le traitement statistique n’est ici pas présent, il semblerait que ces fragments de chromatine 5 et 6 interagissent au sein du noyau contrairement à 6 et 13.
V) Interprétation des résultats Les premiers résultats de l’utilisation de la méthode obtenus par l’équipe de Dekker sont présentés ci dessous :
Figure 3, 4 : Tableau des interactions de la chromatine (Dekker et al, 2002), Représentation 3D du chromosome III de S.cerevisiae (Dekker et al, 2002). Le tableau (Fig.3) présente les distances, en nm, entre les différents segments de chromatine. La proximité des segments est illustrée par des cases de couleurs. L’intensité est d’autant plus élevée que les segments sont proches. La diagonale descendante (de gauche à droite) montre que la distance des fragments successifs est faible : elle illustre le fait que plus deux segments sont proches en nombre de paires de bases sur le chromosome, plus elles sont proches spatialement. La diagonale ascendante révèle la proximité de fragments non successifs, mais qui sont proches spatialement : cela est interprété par la formation de boucles et repliements de la chromatine. La représentation 3D de la chromatine (Fig.4) permet une visualisation claire de la conformation de la chromatine. Il s’agit ici du chromosome III de la levure du boulanger Saccharomyces cerevisiae, obtenu en moyennant les conformations recueillies sur une population de levures.
VI) Intérêts et limites L’avantage majeur de la méthode 3C est qu’elle récupère l’information de conformation de la chromatine in vivo, c’est-à-dire que les produits d’analyses obtenus proviennent directement des cellules vivantes. De plus, elle assure l’obtention de cartes d’organisation spatiale de la chromatine. Elle renseigne ainsi sur les éventuels réarrangements du génome, sans avoir besoin de réaliser un fractionnement cellulaire ou de chromatine. La préparation de l’expérience requiert néanmoins un travail laborieux et chronophage. Il s’agit de la partie critique de la procédure, puisque la résolution et la sensibilité de la technique dépendent en grande partie de sa conception expérimentale. De nouvelles techniques basées sur la 3C ont émergé, chacune ayant des particularités et des applications différentes : La Capture de Conformation Chromosomique Circulaire (4C) est la suite de la méthode 3C. Elle permet de déterminer la nature des séquences en interaction avec une région chromosomique précise, sans présumer de leur identité alors que la méthode 3C consistait à vérifier des interactions prédites ou connues. Enfin, la Capture de la Conformation Chromosomique en Copie de Carbone (5C), permet quant à elle, d’étudier le génome entier (Umlauf, 2015). Par ailleurs, il existe des variantes de la méthode 3C qui sont les méthodes 3C Hi-C et 3C ChiA-PET.
VII) Références bibliographiques
Job Dekker, Karsten Rippe, Martijn Dekker, Nancy Kleckner. (2002). Capturing chromosome conformation. Science. 295, 1306-11. Rahel Gerosa. (2012). Chromosome Conformation Capture. http://www.rna..uzh.ch/dam/jcr:ffffffff-b34e-2810-0000-0000308a3cc2/JC20121106.pdf
Hélène Hagège, Petra Klous, Caroline Braem, Erik Splinter, Job Dekker, Guy Cathala, Wouter de Laat & Thierry Forné. (2007). Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nature protocols. 2,1722 – 1733.
Benoît Moindrot. (2012). Organisation de la chromatine et son lien avec la réplication de l’ADN. https://tel.archives-ouvertes.fr/tel-00733254/document.
David Umlauf. (2015). Le génome intime…et en trois dimensions. Medecine Sciences. 31, 304-311.
Agnès Vernet. (2013). Dans l’intimité chromosomique d’une cellule. Biofutur. http://www.biofutur.com/Dans-l-intimite-chromosomique-d-une-cellule.
| ||




CPG - CHROMATOGRAPHIE EN PHASE GAZEUSE | ||
|---|---|---|
Chromatographie en Phase Gazeuse (CPG) I) Objectifs La chromatographie en phase gazeuse (CPG) a pour but, comme toutes les techniques de chromatographie, de séparer les différentes molécules composant un mélange. Cela permet de connaitre la composition de ce dernier. Elle est surtout utilisée pour des molécules gazeuses ou susceptibles d’être vaporisées par chauffage sans risque de décomposition. II) Principe Le mélange à analyser est vaporisé et mélangé à un gaz porteur à l’entrée d’une colonne de longueur variable, faite d’un capillaire de différents matériaux (silice, acier…), et renfermant une phase stationnaire liquide ou solide. Le gaz porteur ne doit pas présenter d’affinité avec les matériaux constituant la colonne. Les molécules composant le mélange vont traverser la colonne à une vitesse dépendant de leur affinité pour la phase stationnaire. Une analyse est réalisée en sortie de colonne et permet donc de connaitre la composition du mélange. III) Mode opératoire La première étape consiste à purifier le mélange à analyser1. Par exemple, dans le cas de l’analyse d’acides gras, ceux-ci sont trans-estérifiés sur du n-butanol. Il est possible d’ajouter un étalon dans le mélange. Il s’agit d’une quantité précise et connue d’un composé qui n’est pas présent dans le mélange : Par exemple, dans le cas d’une analyse d’acides gras extraits de tissus animaux, il est possible d’ajouter un acide gras à nombre impair de carbones (qui est absent des échantillons, sauf exception). On parle alors d’étalon inerte1. Une quantité d’échantillon allant de 0,2 à 5 µl est injectée dans une chambre en amont du capillaire : l’injecteur. L’injecteur est porté à la température de volatilité de l’échantillon (température légèrement supérieure à la température d’ébullition du mélange). Le mélange gazeux est alors injecté dans la colonne de chromatographie, où les différents composés seront séparés les uns des autres1 A la sortie de colonne, les composés entrent en contact avec le détecteur. Celui-ci évalue la quantité de chaque composé en fonction des modifications des propriétés physiques du mélange gazeux. Il convertit cette modification en un signal électrique envoyé vers un enregistreur. L’enregistreur était autrefois une imprimante qui dessinait directement les résultats sur papier. Il s’agit aujourd’hui d’un ordinateur, qui est en plus capable d’analyser ces derniers1. Les principaux capteurs sont : - le TCD (Thermal Conductivity Detector) qui fait appel à la différence de conductivité des composés. Il utilise pour cela un pont de Wheatstone, faisant varier la tension aux bornes d’un voltmètre en fonction de la conductivité du gaz1. -le FID (Flame Ionization Detector) qui détecte l’ionisation créée par une flamme. L’ionisation modifiant la conductivité du gaz, celui-ci est soumis à une tension constante et l’intensité qui traverse ce gaz est mesurée. -l’ECD (Electron-Capture Detector) qui détecte l’absorption électronique du gaz. Le gaz est soumis à un bombardement d’électrons et un détecteur capte ceux qui ne sont pas absorbés1. -le MS (Mass Spectrometry) qui est basé sur la spectrométrie de masse1. IV) Présentation des résultats Le chromatographe permet, dans un premier temps, l’obtention d’une courbe d’intensité du signal en fonction du temps. Cette courbe est appelée chromatogramme. La courbe présente des pics correspondant aux différents composés présents dans le mélange. Le temps auquel un pic apparait sur le chromatogramme est appelé temps de rétention.
Figure 1 : Exemple d’un chromatogramme obtenu pour un mélange d’acide gras issu de tissu animal. Il est à noter que l’étalon inerte est présent ici avec le C17:0. La surface présente sous le pic d’un des composés du mélange est proportionnelle à la quantité de ce composé dans le mélange. Soit « à la main », soit par ordinateur (plus courant aujourd’hui), il est possible de calculer, par intégration, la surface de chaque pic et sa proportion dans le mélange1. V) Interprétation des résultats La connaissance des temps de rétention permet de décrire qualitativement la présence de certains composés dans le mélange en fonction des pics du chromatogramme. L’analyse du tableau d’intégration permet également de calculer la proportion des différents composés au sein d’un même échantillon. La proportion est égale à la surface du pic du composé d’intérêt divisée par la surface totale sous la courbe1. L’utilisation d’un étalon permet également de calculer la quantité de chaque composé présent dans le mélange. Il est ainsi possible de comparer plusieurs échantillons entre eux1. VI) Intérêts et limites La CPG s’adapte à l’analyse d’un mélange de différents composés. En cancérologie, elle est par exemple utilisée pour identifier des marqueurs de cancer de l’estomac2. En parfumerie, elle sert à dissocier les composants odoriférants d’un élément3. Elle peut également servir pour l’analyse de composés lipidiques, par exemple l’analyse des acides gras composant un tissu, ou celle de composés stéroïdes4. Enfin, elle permet de distinguer des acides aminés. Il s’agit d’un type d’analyse fine : des résultats peuvent être obtenus même si la molécule d’intérêt est présente en faible quantité (de l’ordre du pg pour les plus précis). Cependant, tous les composés ne s’adaptent pas à cette méthode d’analyse. Comme ils doivent être chauffés, les molécules pouvant être dénaturées par la chaleur ne peuvent pas être utilisées. De plus, il faut qu’il n’y ait pas de réaction avec le solvant ou la colonne.
VII) Références bibliographiques 1- Kamoun, P. (1997). Appareils et méthodes en biochimie & biologie moléculaire. Paris: Flammarion.Médecine-sciences.
2- Song H, Peng JS, Dong-Sheng Y, Yang ZL, Liu HL, Zeng YK, Shi XP, Lu BY. (2012). “Serum Metabolic Profiling of Human Gastric Cancer Based on Gas Chromatography/mass Spectrometry”. Brazilian Journal of Medical and Biological Research. 45 (1): 78–85.
3-Brattoli, M., Cisternino E., Dambruoso P., de Gennaro G., Giungato P., Mazzone A., Palmisani J., and Tutino M..(2013). “Gas Chromatography Analysis with Olfactometric Detection (GC-O) as a Useful Methodology for Chemical Characterization of Odorous Compounds.” Sensors. 13 (12): 16759–800.
4- McDonald, J.G., MatthewS., and AuchusR.J.(2011). “Steroid Profiling by Gas Chromatography–Mass Spectrometry and High Performance Liquid Chromatography–Mass Spectrometry for Adrenal Diseases.”Hormones and Cancer. 2 (6): 324–32.
| ||

CRISPR-Cas9 | |||
|---|---|---|---|
CRISPR associated protein 9 (Cas9)
I) Objectifs CRISPR-Cas9 est une technique d’édition du génome développée par Emmanuelle Charpentier et Jennifer Doudna à partir de 2012 (4). Actuellement, ce système permet de supprimer une mutation voire un gène entier, d’insérer un nouveau gène dans un génome ou encore de créer des SNPs. Cette technique peut être utilisée pour faciliter un large panel d’applications en génie génétique, en particulier pour la manipulation ciblée du génome et les thérapies géniques.
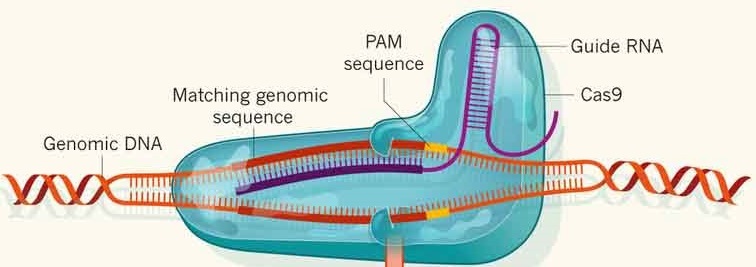
II) Principe La méthode repose sur la capacité de certaines bactéries à résister aux virus grâce aux séquences CRISPR (Clustered Regularly Interspersed Palindromic Repeats), qui sont transcrites en des ARN contenant le complémentaire de l’ADN viral et qui se fixent à la nucléase Cas9. Lorsque ce complexe Cas9-ARN reconnait la séquence complémentaire dans la cellule, il le découpe. C’est cette capacité du complexe à identifier une séquence cible et à la lyser qui fait de CRISPR-Cas9 un outil d’édition précis du génome (3). L’activité nucléase de la protéine Cas9, lui permet d’introduire des Double Stranded Breaks (DSBs), c’est-à-dire des cassures franches sur les deux brins de la molécule d’ADN. Lorsque ces cassures sont repérées par le système de réparation cellulaire, elles sont corrigées de deux manières différentes. D’abord, le Non Homologous End Joining (NHEJ), mécanisme par lequel la cellule insère ou retire de manière aléatoire des nucléotides au niveau de la cassure pour permettre de rabouter les deux extrémités. Ce mécanisme est très imprécis, et tend à provoquer des mutations de type Insertion-Deletion (Indels). L’autre possibilité est la réparation par Homology Directed Repair (HDR), mécanisme par lequel la cellule se base sur une matrice d’ADN de même séquence, qui peut être l’autre allèle du gène concerné, ou bien une matrice d’ADN fournie à la cellule, par exemple sous forme de single-stranded oligonucleotides (ssODNs).
III) Mode opératoire La méthode consiste à fournir à la cellule, soit directement la protéine Cas9, soit sa séquence sous forme d’ADN plasmidique ou d’ARNm. La protéine doit disposer d’un ARN guide (sgRNA), qui est une chimère artificielle entre le crRNA (pour CRISPR) et le tracrRNA (pour trans-activating), et qui lui sert notamment de modèle pour trouver la séquence d’intérêt sur l’ADN de la cellule-cible. Cet ARN peut lui être fourni séparément par transfection dans la cellule-cible, ou bien se trouver sur le même plasmide, sous contrôle d’un promoteur. Cas9 balaie ensuite le génome de la cellule-cible en ne s’arrêtant que lorsqu’elle rencontre un motif correspondant au PAM (Protospacer Adjacent Motif), dont la séquence canonique, présente toutes les 8 bases environ dans le génome humain, est 5’-NGG-3’. Si la séquence d’ADN en amont du PAM correspond à la séquence-cible, Cas9 réalise une cassure double brin entre le 3ième et le 4ième nucléotide en 5’ du 5’-NGG-3’. De nombreuses versions modifiés de cas9 existent, avec une activité nucléase sur un seul brin (nickase), sans activité nucléase (dCas9), avec un PAM modifié (5’-YN-3’), se comportant comme un facteur de transcription, etc... (1, 5)
IV) Présentation des résultats La présentation varie selon le type de Cas9 utilisée et l’objectif de la manipulation. La mesure de l’activité d’une protéine fluorescente peut permettre de mesurer l’efficacité d’une transfection si l’on a transfecté une protéine-chimère, son ARNm ou bien un plasmide. Pour démontrer l’efficacité de la modification d’une séquence cible, on peut réaliser un séquençage type Sanger de cette portion précise du génome. Pour s’assurer qu’aucune autre zone du génome n’a été modifié par erreur (off-target), on pourra soit rechercher les sites ayant une homologie forte avec la séquence cible et les séquencer également, soit faire appel à une méthode de séquençage haut-débit pour séquencer l’ensemble du génome.
V) Interprétation des résultats Dans un premier temps, la mesure d’une activité fluorescente, par exemple, dans la cellule-cible permet de valider la transfection de Cas9. Cela permet d’éliminer les cellules non-transfectées du champ d’étude. Enfin, le résultat d’un séquençage est analysé par alignement avec soit la séquence de départ, soit la séquence souhaitée. La conclusion de cet alignement est binaire : s’il est correct, la modification s’est réalisée avec succès. Dans le cas contraire, la technique n’a pas été efficace.
VI) Intérêts et limites Moins coûteuse et plus rapide que les autres techniques d’édition du génome, CRISPR-Cas9 a été testée sur des bactéries, des cellules végétales, animales et humaines. Le principal intérêt de cette méthode est qu’elle facilement déclinable car il “suffit” de changer la séquence guide du complexe pour l’adapter à une autre édition génomique. Cependant, cette méthode n’est pas fiable à 100% car une séquence complémentaire de la séquence guide pourrait se trouver à un autre endroit du génome et il y aurait donc une mutation sur une autre séquence. Cependant, cette limite n’en est plus vraiment une car une alternative à Cas9 a été développée. En mutant un des deux domaines nucléases de Cas9, les chercheurs ont créé une “nickase” qui ne réalise plus qu’une seule coupure simple-brin. Avec une paire de nickases, chacun ayant une séquence guide correspondant à un des brins, la spécificité de la mutation est améliorée (6). Enfin, des limites éthiques à cette méthode en pleine expansion commencent à apparaitre et doivent être discutées. En effet, la manipulation des cellules somatiques humaines devient plus accessible, avec des risques d’eugénisme en conséquence. De plus les mutations réalisées ne sont pas identifiables par un tiers contrairement aux techniques actuelles. Le caractère génétiquement modifié d’un organisme n’est donc plus vérifiable.
VII) Références bibliographiques 1. Bolukbasi, M. F., Gupta, A. & Wolfe, S. A. (2015) Creating and evaluating accurate CRISPR-Cas9 scalpels for genomic surgery. Nature Methods 13, 41–50
2. Charpentier, E. & Doudna, J. A. (2013) Biotechnology: Rewriting a genome. Nature 495, 50–51
3. Hsu, P. D., Lander, E. S. & Zhang, F. (2014) Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 157, 1262–1278
4. Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna J.A., Charpentier, E. (2012) A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 337, 816–821
5. Perez-Pinera, P., Kocak, D. D., Vockley, C.M., Adler, A. F., Kabadi, A. M., Polstein, L. R., Thakore, P. I., Glass, K. A., Ousterout, D. G., Leong, K. W., Guilak, F., Crawford, G. E., Reddy, T. E, & Gersbach, C. A. (2013) RNA-guided gene activation by CRISPR-Cas9–based transcription factors. Nature Methods 10, 973–976
6. Ran, F.A., Hsu, P.D., Lin, C.-Y., Gootenberg, J.S., Konermann, S., Trevino, A.E., Scott, D.A., Inoue, A., Matoba, S., Zhang, Y., Zhang, F. (2013) Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity.Cell 154, 1380–1389 | |||
CRISPR/Cas9 SAM | ||
|---|---|---|
CRISPR/Cas9 SAM
• Objectifs La méthode CRISPR/ Cas 9 SAM (Synergic Activation Mediator) permet l’activation de la transcription de gènes cibles (Dekeyzer et al., 2018). Cette surexpression a souvent pour objectif de reprogrammer des cellules (Wang et al., 2020), d’étudier la fonction d’un gène, ou encore d’identifier des facteurs de restriction antiviraux (Dekeyzer et al., 2018). • Principe Cet outil, dérivé du système CRISPR/Cas9 bactérien, cible la région promotrice d’un gène à l’aide d’un ARN guide et active triplement sa transcription, c’est-à-dire que l'activation de la transcription est permise par l'action synergique de trois protéines différentes (Dekeyzer et al., 2018).
Figure 1 : Le complexe activateur du système CRISPR/Cas9 SAM : il est composé de dCAS9 en rouge, des ARN aptamères en bleu et des divers activateurs de la transcription (VP64, MS2, p65 et HSF1 en vert) (Dekeyzer et al., 2018) Le complexe utilisé est composé de trois éléments principaux : l’enzyme Cas9 désactivée, un ARN aptamère et une protéine de fusion. La protéine Cas9 (privée de son activité nucléase) ne brise plus les liaisons phosphodiesters entre nucléotides. Elle est fusionnée avec un activateur transcriptionnel (VP64). L’ARN guide possède une séquence complémentaire à la région du génome étudiée et permet l’association avec une protéine de fusion. En effet, cette protéine regroupe la séquence d’adressage de MS2 (protéine d’enveloppe du bactériophage MS2), ainsi que les domaines activateurs de transcription des protéines p65 et HSF1. Le domaine d’activation de MS2 reconnait l’ARN. Les actions coordonnées des protéines VP64, p65 et HSF1 entraînent une triple activation de la transcription du gène cible. L’ARN guide permet donc l’adressage du complexe au niveau du promoteur du gène étudié, et ainsi l’activation de la transcription de ce gène (Dekeyzer et al., 2018). L’application de cette méthode se fait en plusieurs étapes. En effet, il faut fournir le complexe de protéines aux cellules à étudier. Pour cela, il faut construire, d’une part un plasmide contenant la séquence codant pour le (ou les) ARN(s) guide(s) (ou ARNsg), et d’autre part, un ou des plasmides contenant les séquences codant pour Cas9, VP64 et MS2 (Konermann et al., 2015). Prenons l’exemple de l’identification des facteurs de restriction antiviraux. Une des présentations possibles est la mesure de l’activité d’une protéine fluorescente. Le gène d’une protéine fluorescente est introduit dans des cellules. L’expression de ce gène est possible uniquement lorsque le virus est présent (des recombinases excisent alors un codon STOP du gène de la protéine fluorescente). Il est alors possible de visualiser les cellules infectées (fluorescentes) et donc de sélectionner les cellules non-infectées. Ces dernières ont acquis une résistance à l’infection en réponse à la surexpression d’un gène par CRISPR/Cas9 SAM (Heaton et al., 2017).
Figure 2 : Microscopie des cellules exprimant des ARNsg spécifiques de B4GALNT2. Les cellules fluorescentes sont les cellules infectées par le virus. Le pourcentage en blanc est le pourcentage d’infection par rapport au contrôle (Heaton et al., 2017). Reprenons l’exemple précédent. Dans cette étude, la quantification de la fluorescence confirme que la surexpression du gène B4GALTN2 limite l’infection par le virus étudié, par rapport au témoin sans surexpression. CRISPR/Cas9 SAM a donc permis d’étudier le rôle d’un gène et d’identifier un facteur de restriction du virus étudié (Heaton et al., 2017). Le système CRISPR/Cas9 SAM est très robuste et spécifique. Il permet l’activation de la transcription de nombreux gènes, quand il est associé à un pool conséquent d’ARNsg. Les intérêts principaux de cette méthode sont l’étude des gènes codant des protéines ou non, et l’étude de l’organisation du génome, et surtout de sa régulation (Konermann et al., 2015). | ||


CRISPRi Knockdown | |||
|---|---|---|---|
Crispri knockdown
Objectifs CRISPRi (Clustered Regularly Interspaced Short Palindromic interference) est une méthode permettant d’inhiber l’expression d’un ou plusieurs gènes, utilisant dCas9, une version mutée de Cas9, dépourvue de l’activité nucléase1. La force de l’inhibition du gène peut être choisie en modulant certains facteurs, ce qui fait de cette technique un outil intéressant.
Principe Il est possible de créer par assemblage sa propre matrice CRISPR, contenant des séquences codant pour des ARN guides voulus. L’ARN guide transcrit forme un complexe avec dCas9 qui cible le gène d’intérêt. L’enzyme se fixe sur l’ADN cible, mais, à la différence d’un CRISPR « classique », elle ne le coupe pas, mais empêche la fixation optimale de l’ARN polymérase. Le gène ciblé se retrouve donc inhibé. Il existe dans cette méthode différents facteurs contrôlant la force de l’inhibition d’un gène cible, tels que le promoteur de dCas9, le promoteur qui transcrit la matrice, ainsi que le nombre de répétitions dans la matrice de la séquence codant pour l’ARN guide. Il est donc possible de choisir les modalités voulues pour chaque facteur dans le but de contrôler l’intensité de l’inhibition, puis de créer un plasmide avec les séquences citées au-dessus qui sera transmis par transformation à une bactérie pour inhiber les gènes d’intérêts.
Figure 2 : Force des promoteurs pour transcrire la matrice CRISPR2
CRISPRi permet d’inhiber un ou plusieurs gènes et ce jusqu’à 99,9 %. https://doi.org/10.1016/j.cell.2013.02.022.
2 Taylor, George M., Pawel M. Mordaka, and John T. Heap. (2019). ‘Start-Stop Assembly: A Functionally Scarless DNA Assembly System Optimised for Metabolic Engineering.’ Nucleic Acids Research, Volume 47, Issue 3, Page e17, https://doi.org/10.1093/nar/gky1182
| |||




Cryo EM | |||
|---|---|---|---|
Cryomicroscopie Électronique
I) Objectifs La biologie structurale s'efforce de construire des modèles, à terme à une résolution atomique, qui représentent des instantanés de macromolécules biologiques et de décrire la manière dont ces molécules se déplacent. Jusqu'à présent, les structures atomiques n'étaient accessibles que par cristallographie aux rayons X ou par résonance magnétique nucléaire (RMN). Au cours des dernières décennies, ces méthodes ont fourni les principales connaissances sur la structure et la dynamique des macromolécules qui composent les cellules. Toutefois, des défis considérables demeurent. Les deux approches nécessitent des quantités considérables de matériaux, qui doivent être purs et homogènes. De plus, les grands assemblages macromoléculaires (> 300 kD) sont souvent difficiles à étudier à l’aide de ces deux techniques. Or, jusqu'à récemment, la microscopie électronique se limitait à une résolution inférieure, sauf dans les cas particuliers de réseaux bidimensionnels ou hélicoïdaux réguliers. Cependant, de nouveaux progrès techniques se sont réunis au cours des dernières années pour créer une situation dans laquelle les structures atomiques peuvent désormais être déterminées pour des particules de masse moléculaire supérieure à environ 150 kDa. En effet, la cryomicroscopie électronique à particule unique (cryo-EM) offre une approche alternative pour les grosses protéines et les complexes macromoléculaires. Pour la cryo-EM, les macromolécules difficiles à travailler peuvent être purifiées à partir de sources naturelles jusqu'à une pureté et une quantité nettement inférieure à celles requises pour la cristallographie aux rayons X ou la spectroscopie RMN [1,2]. Cette méthode nouvelle de préparation d’échantillons s’affranchissait ainsi de l’utilisation de colorants (par exemple les sels de métaux lourds tels que l’acétate d’uranyle) et des artefacts qui leur sont associés. Ce progrès dans la préparation des échantillons transforma la MET en cryo-MET [3]. Les résultats attendus sont des images à haute résolution de la structure des échantillons congelés, des informations sur la morphologie, la topologie et la dynamique des biomolécules ou des structures biologiques étudiées. Mais aussi des connaissances sur la fonction et le mécanisme d'action des protéines, des virus et des données permettant de reconstruire des modèles tridimensionnels précis des échantillons. Afin d’obtenir à plus long terme, des données cruciales pour la conception de médicaments et la compréhension des processus biologiques fondamentaux. II) Principe En cryo-EM, les assemblages purifiés sont étalés sur un substrat de support et congelés rapidement, produisant un mince film de glace vitreuse contenant les molécules d'intérêt dans des orientations aléatoires. Le processus de congélation rapide permet de garantir que les structures natives des molécules soient largement préservées. En effet, la cryo-EM vise à visualiser des échantillons biologiques congelés, ce qui permet de préserver leur structure naturelle et leur dynamique. Contrairement aux méthodes de microscopies conventionnelles, qui peuvent nécessiter des étapes de fixation, de coloration et de séchage, la cryomicroscopie-EM permet d'observer les échantillons dans des conditions aussi proches que possible de leur état biologique natif [2]. III) Mode opératoire L'échantillon biologique est préparé sous forme liquide, puis congelé rapidement, plongé dans un liquide cryogénique, tel que l'éthane liquide ou l'azote liquide, afin de former un état vitreux sans cristallisation. Ce processus est appelé vitrification et il maintient la structure de l'échantillon intacte à des températures très basses. Une mince couche de l'échantillon congelé est ensuite déposée sur une grille et observée dans le microscope électronique cryogénique, qui est maintenu à une température très basse pour éviter que l'échantillon ne dégèle pendant l'imagerie. Des images sont ensuite acquises à l'aide d'un faisceau d'électrons de 200 kV à 300 kV, permettant ensuite de déterminer la structure des macromolécules observées. Les images de projection proviennent en fait des multiples macromolécules biologiques possédant des orientations aléatoires, en suspension dans le film vitrifié. La structure en trois dimensions de ces macromolécules est donc ensuite déterminée en combinant et en reconstruisant les images de projection dans un volume tridimensionnel (Figure 1 moitié supérieure). Parce que l’information donnée par chaque image est bruyante et incomplète, des dizaines, voire des centaines, de milliers de particules imagées doivent être alignées et moyennées. La qualité de la carte finale de la macromolécule dépend ainsi de la précision avec laquelle les orientations des particules sont déterminées les unes par rapport aux autres par des logiciels ad hoc [3].
Figure 1. Présentation schématique de la reconstruction dans les trois dimensions de particules isolées [3] IV) Présentation des résultats La méthode de cryo-microscopie électronique (cryo-EM) permet d'obtenir des images de structures biologiques à l'échelle moléculaire, offrant ainsi une visualisation détaillée de leur arrangement tridimensionnel. Les résultats de cette méthode sont généralement présentés sous forme d'images de haute résolution des complexes moléculaires étudiés, mettant en évidence leur structure et leur organisation spatiale. Par exemple, il est difficile d’obtenir des images complètes des récepteurs GABA A, un neurotransmetteur, en tant que protéines membranaires. La cryo-EM a permis d'obtenir des structures du GABA A [4]. La cryo-EM permet de visualiser ces interactions à l'échelle atomique en gelant rapidement les échantillons dans des conditions physiologiques. En combinant les images 2D collectées à différents angles, elle permet de reconstruire des cartes de densité 3D des récepteurs GABA A et de leurs complexes avec les ligands.
Figure 2. Structure obtenue à partir d’une cryo EM, après traitement des images en 2D, de deux isoformes différentes du récepteur GABA A humain [4] Les structures du récepteur sont montrées en complexe avec trois ligands : GABA (acide gamma-aminobutyrique) est représenté en rouge, diazépam (une benzodiazépine) est représenté en vert, flumazénil (un antagoniste des benzodiazépines) est représenté en jaune. Les représentations B et C sont des vues de dessus des récepteurs tandis que la vue A est de côté. V) Interprétation des résultats Les résultats de la cryo-EM peuvent être interprétés de diverses manières en fonction des objectifs de l'étude. Par exemple, la résolution élevée des images obtenues permet d'identifier les interactions moléculaires et de comprendre les mécanismes biologiques sous-jacents. De plus, la comparaison des structures moléculaires dans différents états ou conditions expérimentales peut fournir des informations cruciales sur les changements conformationnels et fonctionnels des complexes biomoléculaires. VI) Intérêts et limites La Cryo-EM permet de réduire les dommages d’irradiation causés par le faisceau d’électrons. Elle permet également de préserver la morphologie et la structure des échantillons [5], car le complexe macromoléculaire peut rester dans un environnement proche de ses conditions physiologiques. Aucun colorant ou fixateur chimique n’est utilisé. De plus, la Cryo-EM permet d’obtenir des résultats similaires, voire supérieurs, à ceux de la cristallographie aux rayons X pour la résolution de structures 3D d’objets biologiques. Elle permet également de traiter de lourdes molécules (>300 Kd). Le processus de congélation rapide de l’échantillon, nécessaire pour la cryo EM, est complexe. De plus, ses résultats sont limités pour les petites molécules (<50 Kd). VII) Références bibliographiques [1] CROWTHER R. A. (2016). The Resolution Revolution: Recent Advances In cryoEM. Elsevier Science. Volume 579. [2] MAZHAB-JAFARI M. T. et RUBINSTEIN J. L. (2016). Cryo-EM studies of the structure and dynamics of vacuolar-type ATPases. Science Advances. Volume 2 (n° 7), 1-9. [3] BOUTIN J. A., LI Z., VUILLARD L. et VÉNIEN-BRYAN C. (2016). La cryo-microscopie, une alternative à la cristallographie aux rayons X ? médecine/sciences. Volume 32 (n° 8‑9), 758‑767. [4] TAIANA MAIA O. (2021). Cryo-EM: The Resolution Revolution and Drug Discovery. SLAS Discovery. Volume 26, 17-31. [5] WANG HW. (2015) Cryo-electron microscopy for structural biology: current status and future perspectives. Sci China Life Sci. Volume 58, 750–756. | |||


Cytométrie de Flux (Flow Cytometry) | ||
|---|---|---|
1. ObjectifLa cytométrie de flux est une technique permettant la mesure de multiples caractéristiques physiques et chimiques de cellules biologiques dans des populations parfois hétérogènes et à l’échelle d’une seule cellule. Les applications de la cytométrie de flux sont très variées : immuno-phénotypage, analyse de ploïdie, dénombrement de cellules, quantification de l’émission de GFP (Green Fluorescent Protein)…
2. PrincipeLe cytomètre de flux réalise ces analyses en faisant passer une à une les cellules (jusqu’à plusieurs millier par seconde) à travers un faisceau laser et en capturant la lumière émise par chaque cellule en réponse. L’analyse statistique des données permet de reporter des caractères cellulaires comme la taille, la complexité, le phénotype, et l’état de santé d’une cellule par mesure de la lumière diffusée et de la fluorescence.
3. Mode opératoire
Figure 1 : Présentation du fonctionnement d'un Cytomètre de Flux (http://media.invitrogen.com.edgesuite.net/tutorials/1Intro/player.html)
Voici une représentation simplifiée du cytomètre de flux. Les cellules passent une à une dans un tube et sont soumises à un laser. Plusieurs détecteurs interviennnent alors pour capter les informations relatives à la couleur de la lumière ré-émise par la cellule, à sa taille et à sa granulométrie et sa strucutre. Lorsqu’une cellule passe à travers le laser, elle réfracte ou diffuse la lumière dans toutes les directions. Toutes les intensités détectées sont converties en voltage. La quantité de lumière diffusée vers l’avant ou de faible angle lorsque le laser atteint la cellule est appelé « forward scatter ». Elle est proportionnelle à la taille de la cellule. La quantité de lumière diffusée dans les grands angles lorsque le laser atteint la cellule est appelée « side scatter ». Elle est proportionnelle à la granulosité de la cellule. Le « side scatter » permet donc de déterminer la complexité de la population, car des types de cellule différents auront une structure donc une granulosité différente. Afin d’étudier d’autres caractéristiques cellulaires comme la survie, ou la sous-catégorie cellulaire à laquelle appartient la cellule, des anticorps labélisés avec de la fluorescence sont utilisés. Ainsi, dans la population hétérogène étudiée, certaines cellules seront plus lumineuses que d’autres en fonction de la caractéristique cellulaire spécifique du fluorochrome utilisé. La fluorescence détectée est transmise au détecteur approprié suivant sa longueur d’onde. Là, elle est convertie en une amplitude de voltage proportionnel à la quantité de fluorescence émise. Plusieurs fluorochromes peuvent être utilisés en même temps à condition qu’ils soient compatibles, c’est-à-dire qu’ils aient une longueur d’onde d’excitation semblable mais une longueur d’onde démission différente. On s’affranchit des chevauchements spectraux via la compensation réalisée en tout début d’expérience. Pour ce faire, on réalise une mesure avec des cellules marquées d’une seule couleur et ce pour chaque fluorochrome utilisé. Le logiciel du cytomètre peut alors soustraire pour un fluorochrome la fluorescence provenant d’un autre fluorochrome. La technique du FRET entre deux fluorochores est possible : elle permet de bien séparer le spectre d’émission du spectre d’absorption, ou d’utiliser un laser qui n’exciterait normalement pas le fluorochore émettant en dernier.
4. Intérêts/LimitesLa cytométrie de flux est une technique puissante pour l’étude de caractéristiques cellulaires variées à l’échelle d’une seule cellule. C’est cette possibilité d’analyses multiparamétriques qui constitue le véritable intérêt de la cytométrie de flux. Elle est également réalisable dans un grand nombre de domaines. Les études sont toutefois restreintes à des tailles cellulaires comprises entre 1 et 15 micromètres de diamètre. Mais des dispositifs spéciaux et coûteux permettent de s’intéresser à des cellules allant de 0,5 à 100 micromètres.
5. Expression des résultatsLes pulses obtenus doivent être convertis en données numériques. Cela peut être fait de trois manières différentes : on peut mesurer la hauteur du pulse, la surface sous la courbe et sa largeur. Suivant les cytomètres, la hauteur ou la surface est utilisée. La largeur est gardée pour la distinction entre les événements dus à une ou deux cellules. Une fois que les données ont été mesurées, on peut utiliser des histogrammes ou des nuages de points pour les représenter. Pour observer les corrélations entre deux paramètres, les nuages de points sont à privilégier. Ces graphiques peuvent avoir une échelle linéaire ou logarithmique. Pour éviter la compression sur l’axe des populations négatives pour un anticorps, l’échelle logarithmique est privilégiée. En revanche, pour des expériences mesurant la quantité d’ADN dans les cellules ou pour une gamme étroite de valeurs de fluorescence, la représentation linéaire permet de distinguer les différences plus subtiles. Une échelle bi-exponentielle est utilisable : elle est logarithmique du côté droit et linéaire du côté gauche, c’est-à-dire du côté de l’ordonnée.
6. Interprétation des résultatsLa fluorescence permet également de différencier les populations d’un échantillon en fonction de la réponse positive ou négative à un ou plusieurs fluorochrome.
Figure 2 et 3 : Présentation des résultats obtenus et interprétation (http://media.invitrogen.com.edgesuite.net/tutorials/1Intro/player.html)
On détermine alors toutes les caractéristiques physiques et chimiques des cellules qui nous intéressent au sein de la population de départ.
7. BibliographieMoldavan, A. Photo-Electric Technique For The Counting Of Microscopical Cells. Science 1934: 188-189 Davey H, Kell D. Flow cytometry and cell sorting of heterogeneous microbial populations: the importance of single-cell analyses. Microbiol Rev. 1996;60:641-96 Kalejta R, Shenk T, Beavis A. Use of a membrane-localized green fluorescent protein allows simultaneous identification of transfected cells and cell cycle analysis by flow cytometry. Cytometry. 1997;29:286-91 Hwang K, Park C, Jang S, Chi H, Kim D, Lee J, et al. Flow cytometric quantification and immunophenotyping of leukemic stem cells in acute myeloid leukemia. Ann Hematol. 2012;91:1541-6 http://media.invitrogen.com.edgesuite.net/tutorials/1Intro/player.html
| ||


